This judgment was handed down by the judge remotely by circulation to the parties’ representatives by email and release to The National Archives. The date and time for hand-down is deemed to be 10.30am Thursday 16 March 2023
Table of Contents
The Claim.. 4
Representation. 5
Witnesses of fact 5
The Claimant’s witnesses. 5
The Defendant’s witness. 7
The Expert Evidence. 8
The Drug Development Process. 9
The Background to the DA.. 10
The Development Agreement 11
Events after the DA.. 18
The Law.. 25
The Claimant’s submissions in relation to the ambit of Defendant’s obligations under the DA.. 28
The Claimant’s submission in relation to the Implied Terms. 33
The Defendant’s submissions in relation to the ambit of Defendant’s obligations under the DA.. 34
The Defendant’s submission in relation to the implied term.. 38
Discussion and conclusion in relation to the Defendant’s obligations under the DA.. 39
My findings in relation to the implied terms for which the Claimant contends. 42
The List of Issues. 43
Issue 1(i) 43
Issue 1(ii) 43
Issue 1(iii) 44
Issue 1(iv) 44
Issue 1(v) 44
Issue 1(vi) 44
Issue 2. 45
Issue 3. 45
Issue 4. 46
Causation. 46
Issue 5(i) 46
Issue 5(ii) 47
Issue 5(iii) 48
Issue 5(iv) 48
Issue 5(v) 48
Issue 6. 49
Quantum.. 49
Issue 7. 49
The Defendant’s submissions. 51
Losses not incurred by the Claimant 52
Matters alleged to be outside the ambit of the DA.. 53
Other items which fall to be deducted. 53
The Claimant’s submissions. 55
Can the Claimant claim the costs of the Chirogate invoices paid by CompLex?. 56
Discussion and conclusions in relation to the principles on which the quantum to be awarded to the Claimant 60
Conclusion and disposal 62
MR ANDREW HOCHHAUSER KC:
1. The Claimant, SciPharm S.a.r.l, a company incorporated under the laws of the Grand Duchy of Luxembourg, claims damages from the Defendant, Moorfields Eye Hospital NHS Foundation Trust, which at, the material time, as well as owning the renowned eye hospital in London, had a small pharmaceutical manufacturing division, trading as Moorfields Pharmaceuticals, for an alleged breach of a pharmaceutical drug development agreement made on 20 December 2011 (the “DA”), relating to a product called Treprostinil (the “Product”) for the treatment of lung disease. The DA was initially made between the Defendant and an Austrian company by the name of CompLex Vertriebs GmbH (“CompLex”), ultimately owned by the same investors as the Claimant, which transferred its interest therein to the Claimant with effect from 16 November 2012. This was done, with the consent of the Defendant, for the purpose of holding the pharmaceutical drug’s intellectual property rights in Luxembourg. The Claimant was substituted in the DA by novation, although the DA continued in the terms set out in the original document, where the Claimant is referred to as COM, and the Defendant as CMO, an abbreviation for “contract manufacturing organisation”.
2. The principal issues for resolution are:
(1) whether the loss of a good manufacturing practice (“GMP”) licence by the Defendant constituted a breach of the DA; and
(2) if the Defendant was in breach of the DA, what sums (if any) are recoverable by the Claimant as losses flowing from that breach.
3. The detailed issues, approved by the Court, are contained in Annex 1 to the Costs and Case Management Order of Mr Stephen Houseman KC, sitting as a Deputy Judge of the High Court, dated 4 December 2020, which I will consider in turn below.
4. On 28 January 2020, the Defendant’s application for summary judgment under CPR Part 24 on the grounds that the claim stood no real prospect of success came before HHJ Pelling KC. The application failed. The judgment is reported at [2020] EWHC 269 (Comm).
5. The Claimant was represented by Mr Ali Reza Sinai of Counsel and the Defendant was represented by Dr Andrew Lomas of Counsel. I am grateful to them for their helpful, detailed written and oral submissions.
6. The following witnesses of fact gave evidence on behalf of the Claimant:
(1) Dr Georg Michael Strieder (“Dr Strieder”), who is now a retired consultant in GMP and regulatory affairs. At the relevant time, he was employed by an Austrian company, Orpha Trade GmbH (“Orpha Trade”) as Director of Technical Business Development. In that capacity, he was instructed by CompLex to act as a consultant on their behalf to locate a manufacturer for the Product, which they wished to develop and to obtain a Marketing Authorisation Licence (“MAL”), from the Austrian Agency for Health and Food Safety (“AGES”). He gave background to the DA. He assisted in relation to GMP compliance and making the marketing authorisation application in Austria (the “MAA”). He gave his evidence in English, which he spoke fluently;
(2) Mr Michael Hendrikus Martinus Beckers (“Mr Beckers”), the Managing Director of the Claimant;
(3) Ms Regina Schuller (“Ms Schuller”), who when giving evidence was the Head of Partnering Development at Orpha Trade, but at the material time she was employed by Amomed Pharma GmbH (“Amomed”), a wholly-owned subsidiary of CompLex, and after the novation of the DA, she assisted the Claimant with the development of the Product, liaising closely with Diapharm GmbH (“Diapharm”), a regulatory consulting company based in Austria and Germany used by the Claimant, which filed the MAA with AGES on 25 February 2014. She had, however, no direct contract with anyone from the Defendant;
(4) Ms Bianca Tan (“Ms Tan”), who when giving evidence was the Head of Treprostinil and Medical Device Development for Orphan Pharmaceuticals AG, but at the material time was employed by CompLex and assisted the Claimant with the development of the Product after the novation of the DA. She had primary responsibility for the clinical trials. She was not, however, involved in the negotiations leading up to the DA. She gave evidence in relation to the parties’ performance under the DA and provided details of how she prepared the Schedule of Loss, which is now in its fourth iteration, earlier versions having been corrected.
7. I formed the view that each of the witnesses was truthful and was doing their best to assist the court. As Dr Lomas fairly stated at paragraph 13 of his written closing submissions: “No criticism of their honesty is made. They answered questions fairly, and generally avoided adopting the role of advocate for the Claimant”. It must be remembered however, the events in question took place many years ago and furthermore much of the evidence related to events after the DA had been entered into and was of no assistance to its construction.
8. I also bear in mind the dicta of Leggatt J (as he then was) in Gestmin SGPS S.A. v Credit Suisse Limited, Credit Suisse Securities (Europe) Limited [2013] EWHC 3560 (Comm) concerning the reliability of oral evidence based on recollection of events occurring several years ago:
“Whilst everyone knows that memory is fallible, I do not believe that the legal system has sufficiently absorbed the lessons of a century of psychological research into the nature of memory and the unreliability of eyewitness testimony” [15];
“Memory is especially unreliable when it comes to recalling past beliefs. Our memories of past beliefs are revised to make them more consistent with our present beliefs. Studies have also shown that memory is particularly vulnerable to interference and alteration when a person is presented with new information or suggestions about an event in circumstances where his or her memory of it is already weak due to the passage of time” [18];
“Considerable interference with memory is also introduced in civil litigation by the procedure of preparing for trial…The effect of this process is to establish in the mind of the witness the matters recorded in his or her own statement and other written material, whether they be true or false, and to cause the witness's memory of events to be based increasingly on this material and later interpretations of it rather than on the original experience of the events” [20];
“In light of these considerations, the best approach for a judge to adopt in the trial of a commercial case is, in my view, to place little if any reliance at all on witnesses' recollections of what was said in meetings and conversations, and to base factual findings on inferences drawn from the documentary evidence and known probable facts… Above all, it is important to avoid the fallacy of supposing that, because a witness has confidence in his or her recollection and is honest, evidence based on that recollection provides any reliable guide to the truth” [22]
(Emphasis added).
9. I have adopted the approach indicated in paragraph 22 of the Gestmin decision in relation to all the Claimant’s witnesses of fact. Fortunately, in this case a great deal of the relevant evidence consists of the DA, contemporaneous documents, correspondence and emails.
10. There was only one witness of fact, who gave oral evidence on behalf of the Defendant, Mr Richard Macmillan (“Mr MacMillan”), its General Counsel. He assumed that role on 23 September 2019, some days after the Claim Form in this matter was served. He therefore had no direct knowledge of any of the matters which are the subject matter of this claim. His witness statement consisted of a commentary on the witness statements of Dr Strieder, Ms Tan and Mr Beckers. That commentary consisted principally of submissions by reference to documents. He took instructions from Mr Jonathan Wilson, the Defendant’s Chief Financial Officer, but Mr Wilson also had no personal involvement in this matter at the relevant time. I found his evidence of little assistance in the determination of the issues in this case.
11. There was a potentially significant witness, Ms Margaret Beveridge (“Ms Beveridge”). She was called by neither party, although in closing Mr Sinai accepted that the Claimant could have called her. At the time of the hearing, she was a semi-retired business consultant, living in Kilmarnock, Scotland, where she worked from home. She commenced employment with the Defendant on 18 April 2010 as a Business Development Manager. She remained in that role until she was made redundant in June 2015. She played an important role in relation to the discussions which led to the DA and drafted a 5 year plan on the basis that the Defendant would be the commercial manufacturer of the Product.
12. Although Ms Beveridge made a witness statement dated 29 May 2018, it was not revealed to the Defendant until about May 2021, first in correspondence and then exhibited to the fourth witness statement of Stephen Ian Silverman, a partner in the Claimant’s solicitors, BBS Law incorporating ORG Stock Denton LLP, dated 12 May 2021, in relation to a disclosure application made by the Defendant. In paragraph 15 of that statement, she said that it was standard industry practice for the GMP status to be maintained beyond the production of clinical and stability batches for the licence application and “[the Defendant] knew that a requirement for a successful licence application was the maintenance of GMP status”. By an Order of HHJ Pelling KC dated 30 April 2021, the Claimant was ordered to produce attendance notes of certain aspects of conversations with Ms Beveridge, on the basis that there had been selective waiver of privilege in relation to the same. No notice was served by the Claimant under the Civil Evidence Act 1995 and Ms Beveridge’s evidence was not tested by cross-examination. I therefore attach little weight to it, save insofar as its contents were confirmed by other evidence or admitted by the Defendant.
13. At the Costs and Case Management Conference on 4 December 2020, a single joint expert in the field of pharmaceutical drug development was ordered to produce a report dealing with the issues listed at Annex 2, “identifiable from the face of the pleadings” (the “Annex 2 Issues”) of the Order dated 4 December 2020 of Mr Stephen Houseman KC, sitting as a Deputy Judge of the High Court (the “CCMC Order”). The Defendant initially opposed the appointment of an expert and objected to the provision of the witness statements or any other documents, save the pleadings.
14. The parties agreed to appoint Dr Michael John Desmond Gamlen FRPharmS, FRSC, BSc, PhD (“Dr Gamlen”) as the single joint expert and a joint Letter of Instruction was produced dated 16 January 2021. He has worked as a pharmaceutical consultant, having previously had a long career in the field of pharmaceutical development and pharmaceutical outsourcing. He has a familiarity with drug development projects, such as the one in this case, and is an experienced expert witness. However, despite being asked to provide an opinion on the issues in Annex 2, he appears to have misunderstood his task, and initially on 16 February 2021, he provided a report which addressed the issues in Annex 1, which contain those issues I have to decide. On 21 February 2021, he produced a report addressing the Annex 2 Issues. Thereafter, as permitted by the CCMC Order, the parties put a series of questions to Dr Gamlen, and he gave oral evidence at the hearing.
15. Under the terms of the CCMC Order, the only documents Dr Gamlen saw before he gave oral evidence were the pleadings and with the agreement of the parties, the DA and the AGES Day 70 Preliminary Assessment Report (the “Day 70 Report”). The result was that, on at least one occasion, when shown other documentation he changed his response. He also had a tendency to determine issues of interpretation which were my province, rather than his. Generally, however, I found his evidence to be helpful and reliable, once he had the benefit of material which enabled him to give an informed answer, although I do accept the point made on behalf of the Defendant that on several occasions Dr Gamlen was asked to opine on broader questions that fell outside the ambit of his instructions.
16. In his judgment on the Part 24 application, at [3] HHJ Pelling KC referred to the “various highly complex steps that have to be taken in order to validate a pharmaceutical product prior it being submitted for manufacture … there is a highly regulated process which leads to validation. It is only if validation is obtained that the developer of the drug can then proceed to manufacture and sell it.”
17. There are two separate aspects of the drug development process:
(1) manufacture of the drug for clinical trials tested on patients who have consented to participate. The data from these trials is used to create the Investigational Medicinal Product (the “IMP”) and the Investigational Medicinal Product Dossier (the “IMPD”);
(2) in parallel, validation batches of the drug are produced whereby specific data is collected on the manufacturer’s processes and this information is submitted to the market regulator as part of a MAA. It is intended to demonstrate that the proposed manufacturer named in the application form can consistently reproduce the drug in question under the same manufacturing conditions. The marketing authorisation process is decentralised, and each member state grants its own marketing authorisation number. A detailed application of the validation process under a MAA is described in the Guidance produced by the European Medicines Agency. Of particular importance is the fact that validation is specific to the designated manufacturing site; thus, validation data submitted by the Defendant could only be used for the purposes of validating the Defendant’s manufacturing site and processes.
18. At the heart of this dispute is the ambit of the Defendant’s obligations under the DA. The Defendant contends that, properly construed, it was limited to a simple “fill / finish” agreement to produce 12 clinical study batches, which obligation it discharged. The Claimant’s position is that, on the contrary, there were continuing obligations placed upon the Defendant which it could not fulfil because of the loss of its GMP licence. It contends that the parties were to use the data which the Defendant processed and collected from the initial 12 validation batches, in order to submit those processes for validation in the MAA application, test them for stability and administer the batches (as well as future batches) to conclude the clinical trial dossier. The Defendant was therefore in repudiatory breach of both express and implied terms of the DA.
19. Before turning to the terms of the DA, I should add that it is common ground that, whilst it was clearly envisaged that the Defendant would become the manufacturer of the Product, there was no obligation on the Defendant to do so and no supply agreement was ever entered into between the parties. The Defendant submits that this is fatal to the Claimant’s case because each new manufacturer has to undergo a validation process de novo and any change in formulation, batch size or manufacturing process (even by the same manufacturer) would require validation de novo. It contends that without a supply agreement, there was no contractual obligation on the Defendant to manufacture any further Product, to be the Claimant’s manufacturer or to assist with a MAA or otherwise. The Claimant’s case is that the absence of a supply agreement does not detract from its case, which is founded on alleged repudiatory breaches of the DA alone, which, it says, caused the Claimant substantial loss.
20. In about 2011, for reasons which are unclear, the previous manufacturer was unable to continue to manufacture the Product. Consequently CompLex, which was interested in developing it commercially, instructed Orpha Trade to find a new manufacturer for it. As a result, Dr Strieder entered into discussions with Ms Beveridge and Ms Sophia Titus, who was employed by the Claimant as a project manager, to ascertain whether the Defendant would be interested in manufacturing small batches of the Product for CompLex, which was looking for someone to take the third party manufactured active pharmaceutical ingredient (“API”) in order to develop and thereafter manufacture the Product (the “Project”).
21. Dr Strieder’s evidence, which I accept, was that if the application for a MAL was successful, the manufacturer of the Product for the purposes of obtaining the MAL would then become the commercial manufacturer. The discussions with Ms Beveridge proceeded on this basis because she had been recruited as a Business Development Manager to expand into this area. I also accept that, although the Claimant’s initial quotation did not refer to regulatory steps, in the course of discussions, Dr Strieder made it clear that CompLex was interested in instructing a manufacturer to develop the Product for both clinical trials and for validation in relation to the MAA in a range of concentrations and strengths. This was consistent with the desire of both CompLex and the Defendant that the Defendant should become the manufacturer of the Product, following a successful MAL. The manufacture was also intended to include “compassionate use” in situations where a physician had approved use of an unlicensed/unapproved medicine for those patients having a medical need and for which there was no other suitable licensed medicine available.
22. Throughout 2011 discussions between Dr Strieder and Ms Beveridge continued, with the exchange of scope of work proposals and technical submissions to which Ms Titus contributed. Ultimately, on 20 December 2011, the Defendant entered into the DA with CompLex. As stated earlier, on 16 November 2012, the benefit of the DA was transferred from CompLex to the Claimant.
23. The DA is a detailed and professionally drafted technical agreement, containing 16 clauses and seven annexes. The following express terms of the DA were relied upon by one or other, or both, parties and are of importance:
(1) By clause 1, the parties recorded the background to the DA. In particular, by clause 1.4 it is recorded that:
“It is the intention of COM to enter into further supply agreements with COM or affiliated companies of COM once the PRODUCT is successfully registered in at least one of the European member states.”
(2) By clause 2.1, a number of terms are defined including (emphasis added):
“DEVELOPMENT under this agreement shall mean all work necessary to fulfil the demands of the Guideline on the Requirements to the Chemical and Pharmaceutical Quality Documentation Concerning Investigational Medicinal Products in Clinical Trials (October 2006) by the EMEA CHMP and the Notice to Applicants, Volume 2B, incorporating the Common Technical Document (CTD Part 3.2.P) (May 2008) by the European Commission except for chapter 3.2.P.2. The approach in the case of the PRODUCT is a transfer of the production process from a former manufacturer. Nevertheless, all measures to ensure a smooth production of three validation batches - including but not limited to filtration studies, stress tests, analytical method transfer - are an integral part of the DEVELOPMENT to be performed by CMO. The general approach to obtain the sterile PRODUCT is a standard approach in parenteral manufacturing.
The services are referring to the quotations.
PRODUCT shall mean the finished medical product as defined and specified in Annex ./1.
DOSSIER means such package of technical, clinical and chemical information as is necessary for, or useful in connection with, developing the PRODUCT (EMEA Investigational Medicinal Product Dossier (IMPD) and ICH-M4 Common Technical Document - Format) and includes, without limitation, data in support of formulation, analytical methods and stability concerning the PRODUCT in possession of COM and the chemical, pharmaceutical and biological documentation (including any expert report) and any certificate of free sale.”
(3) By clause 3, the subject matter of the DA is set out, including:
a. By clause 3.1 (emphasis added):
“CMO shall develop for COM the PRODUCT as described in Annex ./1 according to the information therefore provided by COM. CMO shall perform the DEVELOPMENT in accordance with the directives given by COM in writing and in accordance to the development plan given to CMO to COM (Annex ./2). The DEVELOPMENT comprises all necessary development steps for the manufacture and the chemical-pharmaceutical part including long-term stability and quality control. Additional subject matters have to be agreed on in writing and signed by both Parties. It is agreed and understood between the Parties that, contingent on the development character of this project, CMO does not assume any responsibility for the successful DEVELOPMENT of the PRODUCT and the regulatory approval of the product.”
b. By clause 3.3:
“CMO shall perform the DEVELOPMENT of the PRODUCT exclusively for COM. The resulting formulation is exclusive as well. If COM fails to achieve market authorisation for the PRODUCT in at least one of the European Union member states within five years after the signature of this agreement the exclusivity shall be terminated.”
(4) By clause 4, various undertakings are given by the Defendant including:
a. By clause 4.1:
“CMO shall perform all DEVELOPMENT under this Agreement according to the state of the art, in accordance with European current Good Manufacturing Practice (European cGMP) and in compliance with all applicable governmental regulations, as defined by COM and provided to CMO.”
b. By clause 4.4:
“CMO shall use all reasonable endeavours to complete the DEVELOPMENT in accordance with the development plan Annex ./2” (emphasis added)
c. By clause 4.5:
“CMO commits to co-operate with COM to achieve Clinical Study Approval and Marketing Authorisation for the PRODUCT. If any competent governmental authority asks for information related to the DEVELOPMENT of the PRODUCT, CMO shall within 15 working days hand over to COM all available information in written form and at no further cost. If required by any governmental authority, CMO will use its best effort to support any activities. All related costs will be charged separately if not originally part of the DEVELOPMENT or caused by mistake by CMO. If further activities are necessary, CMO shall within 10 working days provide a detailed and mandatory timetable by when this work will be completed.”
d. By clause 4.6:
“CMO shall hand over to COM the DOSSIER in relation to the PRODUCT upon completion of the DEVELOPMENT or upon written demand by COM according to the development plan, Annex ./2
The DOSSIER shall under no circumstances be subject to any right of retention by CMO” (emphasis added)
(5) By clause 10.6, the parties agreed an exclusion clause that stated:
“In no event will either PARTY be liable to the other PARTY for any indirect or consequential loss or damages, including without limitation, direct or indirect loss of profits, arising from or in connection with this AGREEMENT and/or any WORK ORDER.”
(6) By clause 12, the parties agreed various provisions as regards to termination including:
a. By clause 12.1
“This Agreement will come into force on the effective date. This Agreement shall remain in effect for the term defined in the development plan (Annex ./2).”
As will be seen, Annex./2 sets out a series of items of work, with illustrative development timelines. Thus, the end date is when all those work items in Annex./2 are completed.
b. By clause 12.3
“Upon the negligently or wilfully caused failure in fulfilling the development plan or any other material breach or default of this Agreement by either party, the other party shall have the right to terminate this Agreement in whole or in relation to parts thereof by giving thirty (30) days prior written notice. Such termination shall become effective immediately unless COM or CMO shall have cured any such breach or default prior to the expiration of the thirty (30) day period referred to above.”
c. By clause 12.4
“Notwithstanding the above clause 12.3, COM shall have the right at any time to terminate this Agreement in whole within thirty (30) days by giving written notice therefore to CMO.”
d. By clause 12.5
“Upon termination, COM agrees to pay CMO for the DEVELOPMENT which has been performed by CMO prior to termination of the Contract and to pay reasonable and evidenced costs relating to the cessation of the DEVELOPMENT, but such costs will in no case exceed the total payments to be paid by COM as defined in Annex 3.”
(7) Clause 16.6 provided that:
“This Agreement embodies the entire understanding of the parties and shall supersede all previous communications, representations or understandings, either oral or written, between the parties relating to the subject matter hereof. Changes in this Agreement (including this phrase) have to be done in writing.” (emphasis added)
(8) There are then a series of Annexes, the most important of which is Annex./2. This sets out the development plan (the “Development Plan”) and defines the DEVELOPMENT services to be undertaken by the Defendant for the IMPD, as well as what is described as “illustrative development timelines” for the same, which stated “Timelines will be agreed between the parties following signature of contract by both parties.”. That timeline expressly refers to the preparation of Module 3 documentation, which is only relevant to manufacturing for commercial exploitation from the site mentioned in the MAA. The timelines were slightly amended subsequently due to delays in API shipment: this amendment was recorded under Annex./7.
(9) The Development Plan in Annex./2 states that master batch records must be prepared in accordance with cGMP requirements, requires the preparation of the process validation protocol, the execution of process validation and the preparation of an analytical validation report. Process validation relates to the preparation of validation batches for a commercial manufacturing licence application. §9.1 of Annex./2 includes stability testing, which is also expressly included in clause 3.1. The Plan in Annex./2 also thereafter requires stability studies of the finished product over 36 months and the preparation of the IMPD for submission of clinical trial phases I-III with a “Production Timeline” of the end March 2012 for the release of three batches of each dose, at which time the stability study was meant to commence. The Development Plan also includes the preparation of Module 3 documentation, which is only relevant to manufacturing for commercial exploitation from the site mentioned therein.
(10) §9.1 of Annex./2 includes stability testing, which is also expressly included in clause 3.1.
(11) The Defendant thereafter drafted a detailed development plan called P-258 (the “Detailed Development Plan”), the contents of which are very similar to those contained in Annex 2, but which develop various of the obligations in further detail and, as envisaged by the opening words of Annex ./2 in relation to timelines, which stated: “Timelines will be agreed between the parties following signature of contract by both parties”, set out specific timelines, assigning responsibility to various departments for particular steps and indicating that “Unless stated, the next step cannot be carried out until the previous one has been completed.” The precise date of the document is unclear. It was suggested by Dr Lomas that it possibly preceded the DA, and therefore was to be treated as a pre-contractual document, excluded by reason of clause 16.6 of the DA. In my judgment, however the wording of the document in §2.0, entitled ‘BACKGROUND’, begins: “Moorfields Pharmaceuticals have been contracted by Orpha Trade to manufacture a medicine for clinical trial supplies.”, makes clear that the DA had already been entered into and therefore has to be regarded as part of the DA, pursuant to the provisions of Annex.2./. I would draw attention to the following aspects of the Detailed Development Plan:
a. The formulations are set out at §2.3;
b. §3.2 is entitled ‘Phase 2 Clinical / process validation batch manufacture’ and provides:
“[the Defendant] will manufacture a 3 batch campaign for each strength. The purpose of these can be seen in Table 5. [The Defendant] will generate a Process Validation protocol. Analytical Development will generate a stability protocol.”
c. Tables 5 to 8 then set out various technical details relating to the manufacture of the validation batches. As set out above, Table 5 defines the purpose of the validation batches as being:
|
Table 5: Phase 2 Testing |
|
|
Purpose |
Testing |
|
Batch 1 |
Stability samples
Process Validation samples
Finished product release samples
Sterility validation samples
Endotoxin validation samples |
Mixing validation, pH adjustment volumes, weight per mL, filter discard (confirmation), fill volumes |
|
Batch 2 |
Stability samples
Process Validation samples
Finished product release samples |
Confirmation of parameters determined above |
|
Batch 3 |
Process Validation samples
Finished product release samples |
Confirmation of parameters determined above |
|
Batch 4 |
Finished product release samples |
Finished product samples |
d. Table 9 details various “design inputs” including:
“Regulatory and statutory requirements of intended markets: This is for clinical trial in Europe and is therefore governed by European legislation”.
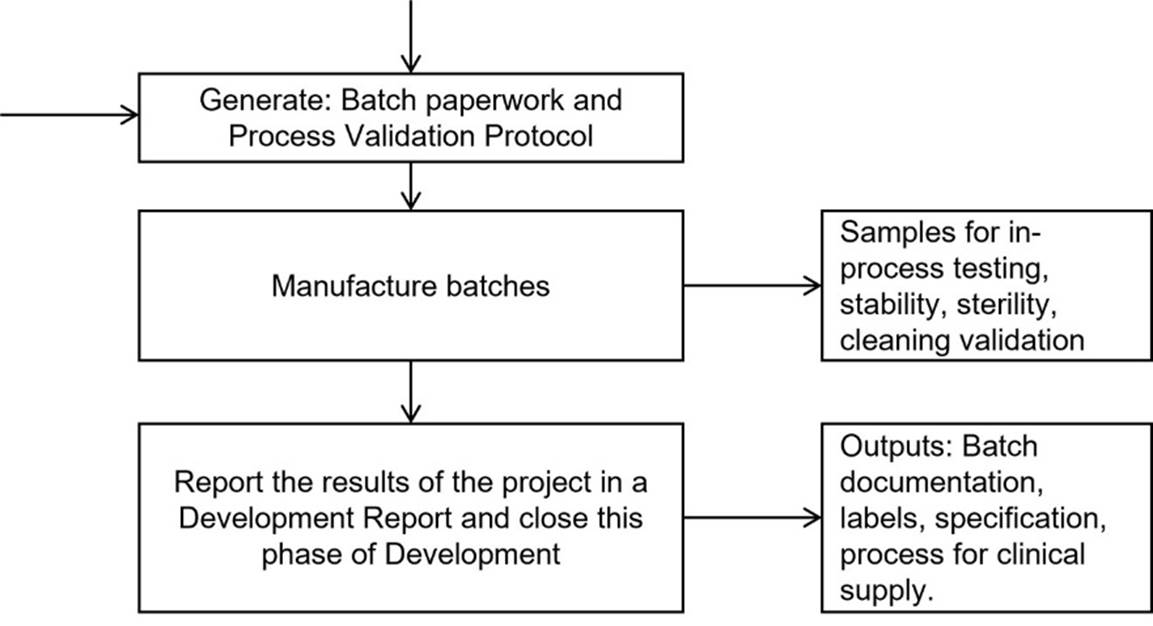
e. Figure 2 turns the Detailed Development Plan into a flow chart, which includes the generation of “Batch paperwork and Process Validation Protocol” and ends with the generation of a “Development Report” (which correlates with Step 12 in Table 11):
The Implied Terms in the DA alleged by the Claimant
24. Whilst acknowledging that, given the terms of the DA, there is limited scope or need for the implication of terms, the Claimant contends that there were also terms to be implied into the DA. Paragraph 25 of the Particulars of Claim formulated the matter in this way:
“The parties’ intention and agreement, as reflected in the express terms of DA, by standard industry practice and by implication for business efficacy and/or to give effect to the parties’ intention was that [the Defendant] would:
(i) develop and manufacture process validation batches of the Drug for the purpose of submitting an application to European regulators for a commercial manufacturing licence. Process validation requires the approval of the manufacturing equipment and methodology and demonstrates that the commercial batch size is consistently reproducible under the validated controlled conditions. The parties’ subsequent conduct is evidence of the contractual intention as it existed when the DA was entered into. The Claimant submitted a manufacturing licence application to the Austrian regulator (referred to in more detail below) which included process validation data prepared by [the Defendant]. This was only required for the commercial manufacture of the Product since there would have been no requirement for validation data in respect of clinical trials to be included within the Claimant’s application, given that a standard sterilisation process was described in the pharmacopoeia;
(ii) develop and manufacture batches of the Drug to be used in clinical trials;
(iii) carry out stability testing on sample batches at regular intervals for up to 36 months;
(iv) be named as the commercial manufacturer in the Claimant’s manufacturing licence application;
(v) continue to develop the Drug until the Claimant’s application was finally determined, although [the Defendant] did not guarantee that the final Product could be successfully manufactured or that a manufacturing licence would be obtained;
(vi) as the commercial manufacturer named in the manufacturing licence application, co-operate with the Claimant at no additional cost in responding to the regulator’s questions and requests for information, and make any changes to the Drug determined necessary by the regulator as part of the licence application process;
(vii) maintain its European current Good Manufacturing Practice (cGMP) accreditation throughout the Development and until final determination of the Claimant’s manufacturing licence application;
(viii) carry out the Development exclusively for the Claimant for at least 5 years, and longer if a manufacturing licence was obtained in that period. In that event, the parties’ intention was for MP to be the licensed commercial manufacturer of the final approved Drug for sale and commercial exploitation;
(ix) prepare the Module 3 documentation for the licence application. MP’s Sophia Titus (described in Annex 6 of the DA as the Project Manager CTM and as a Responsible Person) contacted the Claimant 26 March 2013 to state that Premilla Pillay (described in Annex 6 of the DA as the Regulatory Manager and as a Responsible Person) was leaving and seeking permission to outsource the Module 3 report to an external consultant so that the report could be ready in time for filing the licence application. The Module 3 Report, including the required data for commercial manufacturing in Section 3.2.P.8.2 concerning the Post-Approval Stability Protocol and Stability Commitment, was eventually produced by MP and invoiced thereby to the Claimant on 28 May 2013.”
25. Suffice it to say that the basis upon which these terms fall to be implied and their content is hotly contested by the Defendant.
26. On 7 March 2012, Dr Strieder carried out a GMP audit of the Defendant’s site.
27. On 17 August 2012, the Claimant purchased 150 grams of API (in three batches of 50 grams) from Chirogate International Inc (“Chirogate”), a Taiwanese supplier, for US$932,880 whilst the DA was still in the name of CompLex, and this was shipped directly from Taiwan to the Defendant in London. This API was used to manufacture the 12 validation batches between November 2012 and January 2013, although subsequent batches were manufactured for clinical trials until November 2013, shortly before sterile manufacturing was suspended at the Defendant’s site, following critical inspections by the UK Medicines and Healthcare Products Regulatory Agency (the “MHRA”) starting in December 2013, which identified failures to comply with GMP.
28. By an additional scope of works order dated 1 November 2012, the size of the vials was changed to 20ml, and stability testing was extended to 36 months. It was one of a number of additional scope of works orders, which effectively supplemented Annex./2.
29. On 16 November 2012, the DA was transferred to the Claimant.
30. On 22 November 2012 the Defendant’s Validation Protocol was signed off. This was a validation document for the process validation batches, being three active batches for each concentration of the solution.
31. On 17 January 2013, there was a telephone conference call between Dr Strieder and an Andrew Froome and Sophia Titus on behalf of the Defendant. Amongst other things, the Defendant was informed by Dr Strieder that Orpha Trade /the Claimant wished to file the MAA, accompanied by three months of stability data. If possible, the Claimant wanted to have the first draft of Module 3 by the end of March 2013, with the process validation report.
32. On 28 February 2013, the Defendant produced its Validation Summary Report.
33. On 11 March 2013, additional work was agreed for the generation of the drug product section of Module 3 at a price of £6,573.
34. In November 2013, section 3.2.P.3.1 of CTD Module 3 named the Defendant as the proposed manufacturer and stated that the Product would be manufactured in accordance with GMP.
35. On 19 December 2013, without prior notice to the Claimant, the Defendant voluntarily suspended manufacturing. It appears that the Defendant was unable to carry out aseptic filling and sterile terminalisation in the preparation of the vials containing the Product in accordance with GMP. Although it was frequently stated on behalf of the Defendant that its remedial action plan would result in a successful further audit by the MHRA, and the resumption of manufacturing, this did not happen and, eventually, in January 2015, the Defendant closed its site.
36. Ms Tan’s evidence, which I accept, was that when the Defendant lost its GMP status, the Defendant’s staff with whom she was dealing, including Ms Beveridge, appreciated the serious impact the suspension of manufacturing would have on the Project. There was a need for a continuous supply of the Product under the DA for the clinical trials. Once a patient had been included in the trial and commenced taking the Product, he or she needed to receive the Product for life. An interruption in its supply could have been life-threatening. Immediate emergency measures had to be taken, such as re-labelling existing batches destined for other use and extending the shelf-life of available vials. The re-labelling exercise and liaising with clinical trial centres in different European Union (“EU”) member states took considerable time. On 10 February 2014, Mr Skelton, the Defendant’s Quality Assurance Manager, issued new certificates of analysis for the batches manufactured in November 2013 with their shelf-lives extended to June 2014 to cater for the suspension of manufacturing.
37. As stated at paragraph 6(3) above, on 25 February 2014, Diapharm filed the MAA in Austria in relation to the Product. The Defence challenged the basis on whose behalf that application was made. In my judgment, it was clearly made on behalf of the Claimant, as the email dated 10 September 2014, from Dr Risse, an employee of Diapharm, to Ms Beveridge stated. This was the conclusion of Dr Gamlen, at paragraph 30 of his initial report, addressing the Annex 1 issues, where he stated that it is not unusual for such applications to be filed by local agents to ensure that local requirements are met.
38. On 27 February 2014, the Hungarian regulator confirmed to Dr Risse that frozen samples could be sent directly from the UK to Hungary, and he requested the Defendant to send samples directly to Hungary. This was done. This therefore indicates that the Defendant submitted samples for the purpose of validation in respect of a MAA.
39. At this stage the Defendant had the acquired Development know-how and technology and there was no other way to manufacture clinical trial batches. In order to guarantee a continuous supply of Product to the clinical study and to proceed with the MAA process, a new manufacturer had to be found. There was little available choice of alternative manufacturers.
40. Following a request from AGES, on 13 March 2014 Mr Skelton confirmed that the Defendant was licensed for manufacturing with batch certification.
41. Following inspections on 6 and 7 May 2014, the MHRA wrote to the Defendant notifying it of on-going failures to comply with the GMP.
42. On 28 May 2014, the MHRA agreed that in view of the dangers to patients, previously manufactured Product could continue to be supplied to patients provided certain conditions were satisfied, including confirmation from the Claimant as the sponsor. The MHRA accepted that the timelines for transferring the drug to another manufacturer would not alleviate the potential shortages of supply, however they expressly wrote that reinstatement of manufacture of the Product “is not authorised”. The MHRA expressly requested the parties to provide plans of how the Product could be transferred to an alternative manufacturing site whilst maintaining continuity of supply.
43. The Defendant’s own risk assessment carried out in about mid 2014 stated:
“Moorfields has undertaken a comprehensive search of small scale sterile manufacturers but to date a suitable site has not been found - due to limited capacities, capabilities and the timelines for the transfer of manufacturing…. Moorfield has therefore not been able to identify to date a manufacturer that has the capabilities, plus the ability to commence immediately the transfer of the manufacturer of Treprostinil clinical supplies. Notwithstanding that, even with an immediate start, the continuity of clinical supplies would be compromised, which would result in a medically critical situation….
Patients who have been recruited into clinical study, and who respond positively, are maintained through compassionate supply. This continued compassionate supply is diluting the availability of previously manufactured stock and this reducing the supplies available to continue active recruitment. Active recruitment is required to successfully compete the clinical programme.
Moorfields and SciPharm have worked closely to develop and manage the production process, to complement and satisfy the ongoing clinical requirements. Based on the timelines and assumptions, Moorfields and SciPharm believe that the process to transfer the production elsewhere would inevitably have an impact on the clinical programme. Transfer would not ensure continuity of supply and would result in a medically critical shortage of study medication and compassionate supplies. To ensure continuity of supplies, it will be necessary to utilize the stock already held at Moorfields and for Moorfields to manufacture a further batch.”
44. Unfortunately, the Defendant’s request for permission from the MHRA to manufacture an emergency batch for patients who faced stock shortages was unsuccessful. Recipharm AB, a Swedish manufacturing company based in Solna (“Recipharm”) was identified as the only other available manufacturer worldwide to manufacture Product in the time available before stocks were depleted, but, even with an immediate start, there was still the problem of ensuring continuity of supply. Since no manufacturing had been undertaken by the Defendant since 2013, the result was that there would be a critical shortage of the 10 mg strength of the Product by Q4 2014. It was therefore necessary for the Defendant to manufacture a further batch of the 10 mg strength which could be released in early Q4 2014.
45. The only solution available to achieve this was for Recipharm to manufacture an emergency batch at increased cost, requiring the Defendant urgently to transfer the technology to Recipharm, on the basis that aseptic filling and sterile terminalisation would be undertaken in Sweden, and labelling, packaging and batch release would be done by the Defendant.
46. On 25 June 2014, the Claimant entered into a new development agreement with Recipharm (the “New DA”), in materially the same terms as the DA.
47. On 29 July 2014, AGES produced its Day 70 Report on the MAA. It named the Defendant as the proposed manufacturer for both dosage and batch release. It stated that the MHRA’s statement on the Defendant’s non-compliance with GMP was a “major objection” and they wrote to Diapharm posing 63 technical questions on the drug substance and product before a decision would be taken on the MAA.
48. On 1 August 2014, Dr Strieder sent the Defendant AGES’ list of 63 questions to the Defendant and requested answers by mid-August 2014. On 14 August 2014 the Defendant sent its replies to the 63 questions. In response to question 33, it stated that it, rather than Orpha Trade, should be named as the manufacturer and the role of Orpha Trade should be explained by the Claimant.
49. From the minutes of a meeting held on 30 September 2014 between representatives of the parties, which minutes were amended by Ms Tan and Dr Strieder, it appears to have been agreed that final answers to the questions posed by AGES would be submitted by the first week of December 2014.
50. In October 2014 two anonymous whistleblowing letters were sent to the MHRA, one from “a number of staff”, complaining of bad working practices, poor leadership, staff bullying, resignations and a series of GMP breaches.
51. Pursuant to requests made by the Defendant on 6 and 8 October 2014, on 28 October 2014, the MHRA wrote to the Defendant that they had reviewed the outsourcing proposal and did not object to the technology transfer to Recipharm. The parties interpreted this as permission for Recipharm to manufacture and for the Defendant to pack, label and release the batches.
52. However, on 6 November 2014, Philip Greaves, the Director of Quality wrote to Dr Strohmaier, the Managing Director of the Claimant, and Ms Tan to state that, as a result of the whistleblowers’ complaints, the MHRA had inspected the Defendant on 30 October 2014 and had upheld some of the allegations. He attached a copy of the inspectors’ report and that said the Defendant was waiting for a decision. Mr Greaves stated that until they had heard back from the MHRA, even existing supplies labelled and prepared for dispatch would not be released.
53. On 13 November 2014, the MHRA wrote to the Defendant, giving notice that in view of the deficiencies in relation to GMP inspected on 30 October 2014 and which had been continuing since 19 December 2013, the decision had been taken to suspend that part of the Defendant’s manufacturing licence which authorised aseptic manufacture and terminal sterilisation processes with immediate effect until 12 February 2015. The MHRA wrote that there was a risk to public health arising from a lack of sterility assurance and that undertaking further terminal sterilisation would constitute a criminal offence.
54. In late November 2014, the Defendant audited Recipharm for the technology transfer and the emergency batch was manufactured by Recipharm and released by the Defendant.
55. In response to a request from Dr Strohmaier on 26 January 2015 as to whether the Defendant expected to regain full operational capacity, on 27 and 28 January 2015, the Defendant indicated that it planned to cease all production of pharmaceutical products. An email dated 28 January 2015 from Tim Record, the Defendant’s Operations Manager, pasted a copy of a formal announcement which included the statement: “Given the length of the suspension, the board has concluded that re-instating manufacturing after such a lengthy pause would be too costly and complex.”
56. On 3 February 2015, a telephone conference call took place between Dr Strohmaier, Dr Strieder and Ms Tan on behalf of the Claimant and Mr Record, Mr Froome and Ms Beveridge on behalf of the Defendant, when the Claimant was informed that the Defendant would cease all activities around the end of April 2015, including manufacturing, packaging and stability and would close its production facility. As a result, the Defendant could no longer complete the stability testing on manufactured batches and the Claimant was required to transfer stability testing and packaging to another manufacturer.
57. On 7 May 2015 the Defendant’s then solicitors sent a draft Deed of Termination seeking to terminate the DA and to release the Defendant from all liability. The Claimant did not execute that document.
58. On 15 December 2015 the Claimant sent a letter before claim to the Defendant.
59. In November 2016, Recipharm prepared a new Validation Report and a new Module 3 submission for the Claimant.
60. After a period of postponement, on 14 June 2018, the MAA was reinstated, with Recipharm being named as the commercial manufacturer for compounding, filling, sterilisation, packaging and labelling. It named Amomed as the manufacturer for batch release and the marketing authorisation holder. The MAA was carried through to market authorisation.
61. From the beginning of 2019, marketing authorisations were given in various EU member states in the name of Amomed. The Claimant relied upon the Hungarian licence in respect of the 10 mg solution, as an example of a MAA, which, whilst showing Amomed as the licence holder, expressly states that the “applicant submitted an application for marketing authorisation” on 25 February 2014, this being the original application filed by Diapharm which was stayed and then reinstated with Recipharm as manufacturer.
62. On 13 September 2019 the Claimant issued its claim.
63. I would add, for completeness that the Product is also used to treat Chronic Thromboembolic Pulmonary Hypertension (“CTREPH”). The Claimant filed an entirely separate marketing authorisation application in respect of CTREPH that has no relevance to this claim. There are a number of documents in the bundles which refer to this. That application was withdrawn due to a technicality, was re-submitted and has been authorised since 2020.
64. I turn to the principles to be applied when construing contractual agreements.
65. First, in relation to construction of express terms, there is a helpful summary contained in the judgment of Stuart-Smith LJ, who gave the only reasoned judgment in European Film Bonds A/S v Lotus Holdings [2021] EWCA Civ 807 at [43]-[44], with which Sir Nicholas Patten and Asplin LJ agreed, where he stated:
“43. The Judge, having referred to Rainy Sky SA v Kookmin Bank [2011] UKSC 50, Arnold v Britton [2015] UKSC 361 at [15] and Wood v Capita Insurance Services Ltd [2017] UKSC 24, summarised the relevant principles of contractual construction at [52] as follows:
“(1) The court's task is to ascertain the objective meaning of the language which the parties have chosen to express their agreement. It has long been accepted that this is not a literalist exercise focused solely on a parsing of the wording of the particular clause, but that the court must consider the contract as a whole and, depending on the nature, formality and quality of drafting of the contract, give more or less weight to elements of the wider context in reaching its view as to that objective meaning.
(2) Interpretation is a unitary exercise; where there are rival meanings, the court can give weight to the implications of rival constructions by reaching a view as to which construction is more consistent with business common sense. But, in striking a balance between the indications, given by the language and the implications of the competing constructions, the court must consider the quality of drafting of the clause.
(3) The court must also be alive to the possibility that one side may have agreed to something which with hindsight did not serve his interest. This exercise involves checking each suggested interpretation against the provisions of the contract and investigating its commercial consequences. Similarly, the court must not lose sight of the possibility that a provision may be a negotiated compromise or that the negotiators were not able to agree more precise terms.
(4) Textualism and contextualism are not conflicting paradigms in a battle for exclusive occupation of the field of contractual interpretation. Rather, the lawyer and the judge, when interpreting any contract, can use them as tools to ascertain the objective meaning of the language which the parties have chosen to express their agreement. The extent to which each tool will assist the court in its task will vary according to the circumstances of the particular agreement or agreements.
(5) Account should be taken of the fact that negotiators of complex formal contracts may often not achieve a logical and coherent text because of, for example, the conflicting aims of the parties, failures of communication, differing drafting practices, or deadlines which require the parties to compromise in order to reach agreement. There may often therefore be provisions in a detailed professionally drawn contract which lack clarity and the lawyer or judge in interpreting such provisions may be particularly helped by considering the factual matrix and the purpose of similar provisions in contracts of the same type.
43. The provenance of each element of this statement of principles is clear and uncontroversial. The principles were adopted by the parties for the purposes of the appeal. With one minor gloss, I fully endorse that approach: it is quite unnecessary for the Court to provide yet another iteration of the relevant principles or to cite chunks of the leading authorities which underpin the Judge's formulation. The only gloss that I would apply is to recognise that most iterations of these principles, even at the highest level, have subtle differences of emphasis. It is usually clear that these differences are because the Court will have in mind the facts of the particular case and so may highlight aspects of the general principles that are particularly relevant to the case that it has to decide. That said, I would normally include in any iteration of the principles, the principle derived from ICS v West Bromwich Building Society [1998] 1 WLR 896, 912H, reaffirmed with slight refinements many times since, that interpretation is the ascertainment of the meaning which the document would convey to a reasonable person taking into account facts or circumstances which existed at the time that the contract was made, and which were known or reasonably available to the parties to the contract.”
66. In relation to whether terms fall to be implied into a contract, there was a helpful and uncontroversial summary of the relevant authorities contained in paragraphs 116 to 118 the Defendant’s written submissions as follows:
“116. As for implication of terms, in Philips Electronique Grand Public SA v British Sky Broadcasting Ltd [1995] EMLR 472, 481, Sir Thomas Bingham MR explained that it was “difficult to infer with confidence what the parties must have intended when they have entered into a lengthy and carefully-drafted contract but have omitted to make provision for the matter in issue", because “it may well be doubtful whether the omission was the result of the parties' oversight or of their deliberate decision”, or indeed the parties might suspect that "they are unlikely to agree on what is to happen in a certain ... eventuality" and "may well choose to leave the matter uncovered in their contract in the hope that the eventuality will not occur.
117. Sir Thomas went on to say this at p.482 (emphasis added):
“The question of whether a term should be implied, and if so what, almost inevitably arises after a crisis has been reached in the performance of the contract. So the court comes to the task of implication with the benefit of hindsight, and it is tempting for the court then to fashion a term which will reflect the merits of the situation as they then appear. Tempting, but wrong… [I]t is not enough to show that had the parties foreseen the eventuality which in fact occurred they would have wished to make provision for it, unless it can also be shown either that there was only one contractual solution or that one of several possible solutions would without doubt have been preferred ...”
118. The judgment of the Supreme Court in Marks and Spencer plc v BNP Paribas Securities Services Trust Company (Jersey) Ltd & Anor (Rev 1) [2015] UKSC 72 restated the law at [14] to [32] citing, inter alia, Philips Electronique, to which Lord Neuberger added the following comments:
[21] In my judgment, the judicial observations so far considered represent a clear, consistent and principled approach. It could be dangerous to reformulate the principles, but I would add six comments on the summary given by Lord Simon in BP Refinery as extended by Sir Thomas Bingham in Philips and exemplified in The APJ Priti. First, in Equitable Life Assurance Society v Hyman [2002] 1 AC 408, 459, Lord Steyn rightly observed that the implication of a term was "not critically dependent on proof of an actual intention of the parties" when negotiating the contract. If one approaches the question by reference to what the parties would have agreed, one is not strictly concerned with the hypothetical answer of the actual parties, but with that of notional reasonable people in the position of the parties at the time at which they were contracting. Secondly, a term should not be implied into a detailed commercial contract merely because it appears fair or merely because one considers that the parties would have agreed it if it had been suggested to them. Those are necessary but not sufficient grounds for including a term. However, and thirdly, it is questionable whether Lord Simon's first requirement, reasonableness and equitableness, will usually, if ever, add anything: if a term satisfies the other requirements, it is hard to think that it would not be reasonable and equitable. Fourthly, as Lord Hoffmann I think suggested in Attorney General of Belize v Belize Telecom Ltd [2009] 1 WLR 1988, para 27, although Lord Simon's requirements are otherwise cumulative, I would accept that business necessity and obviousness, his second and third requirements, can be alternatives in the sense that only one of them needs to be satisfied, although I suspect that in practice it would be a rare case where only one of those two requirements would be satisfied. Fifthly, if one approaches the issue by reference to the officious bystander, it is "vital to formulate the question to be posed by [him] with the utmost care", to quote from Lewison, The Interpretation of Contracts 5th ed (2011), para 6.09. Sixthly, necessity for business efficacy involves a value judgment. It is rightly common ground on this appeal that the test is not one of "absolute necessity", not least because the necessity is judged by reference to business efficacy. It may well be that a more helpful way of putting Lord Simon's second requirement is, as suggested by Lord Sumption in argument, that a term can only be implied if, without the term, the contract would lack commercial or practical coherence.
[…]
[29] …the process of implication involves a rather different exercise from that of construction. As Sir Thomas Bingham trenchantly explained in Philips at p 481:
“The courts' usual role in contractual interpretation is, by resolving ambiguities or reconciling apparent inconsistencies, to attribute the true meaning to the language in which the parties themselves have expressed their contract. The implication of contract terms involves a different and altogether more ambitious undertaking: the interpolation of terms to deal with matters for which, ex hypothesi, the parties themselves have made no provision. It is because the implication of terms is so potentially intrusive that the law imposes strict constraints on the exercise of this extraordinary power.”
67. Applying those principles, I turn to the central issues of the ambit of the Defendant’s obligations under the DA. Both Counsel have made lengthy written and oral submissions, before, at and after the hearing. I do not intend to reproduce them at length, but instead produce a summary of the principal points taken. Suffice it to say that I have re-read the transcripts of the hearing and have carefully considered all the parties’ written submissions.
68. In summary the Claimant’s submissions were as follows:
(1) Properly construed, the Defendant’s obligations went further than simply one of “filling and finishing” the manufacture and delivery of the twelve validation batches. The DA set out a long term commitment between the parties to develop the Product through clinical trials and the marketing authorisation process, until the Product, clinically tested on patients and authorised for commercial sales on the market, was developed;
(2) The parties were to use the data which the Defendant processed and collected from the initial 12 validation batches in order to submit those processes for validation in the MAA, stability test those batches over 36 months and administer the batches (as well as future batches) to conclude the Dossier;
(3) Clause 4.1 contained an undertaking from the Defendant to perform “all Development” in accordance with GMP. As Dr Gamlen said: “the requirement for the manufacturer to maintain their GMP status during, and after completion, of the development work is absolute” ;
(4) It is clear that the DA required development for a MAA. Mr Sinai relied upon the following:
(i) In clause 2.1, the definition of “Development” expressly referred to “all work necessary to fulfil the demands of the… Common Technical Document”, which is the MAA. The definition of “Development” excludes the work required for Part 3.2.P of the CTD (which is one sub-part of Module 3 of the application form). Since only this aspect was excluded, the rest of Module 3 was necessarily included in the DA. In addition, Module 3 is expressly included in the Illustrative Development Timelines of Annex 2. Furthermore, nothing turns on the exclusion of Part 3.2.P because the Defendant costed Module 3 in a subsequent scope of work document and the Claimant paid for that work, including Part 3.2.P. It therefore fell within the additional work agreed between the parties envisaged in clause 3.1 of the DA. In the event, the Defendant prepared all of Module 3, which included the data and analytical test reports that the Defendant carried out on the validation batches that it manufactured;
(ii) In clause 2.1, the definition of “Dossier” expressly included information in connection with the “Common Technical Document” and importantly includes “any expert report”. The reference to an expert’s report in the definition of Dossier in the DA is proof that it was for an MAA. As Dr Gamlen stated “All of the documentation that… I have been shown confirms that this was, indeed, preparation for market authorisation” ;
(iii) In clause 3.1, the Development is stated to comprise “long-term stability and quality control” [emphasis added]. The latter is a reference to the MAA;
(iv) Looking at the contract as a whole, the indicia are that the Defendant’s obligation was more than a “fill and finish” obligation:
(a) clause 1.4 refers to a supply agreement “once the product is successfully registered”. Those words must refer to market authorisation;
(b) under clause 3.3 the Defendant agreed to perform the Development of the Product exclusively for the Claimants for 5 years (being the time that it usually takes to bring a drug to the market) in order for the Claimant to “achieve market authorisation” in at least one European member state. Should that not happen, the DA would be terminated;
(c) clause 4.5 provided that the Defendant “commits to co-operate with [the Claimant] to achieve Clinical Study Approval and Marketing Authorisation for the Product.” [emphasis added]. It follows that the on‑going work required for the MAA under clause 4.5 to “achieve market authorisation” is part of the “Development” and therefore must be performed “in accordance with GMP.” This clause expressly included the provision of information, in written format and at no extra cost, to the market regulator. It also included further “activities” if required by the regulator, albeit the Defendant was entitled to be remunerated if such activities were not initially costed. As stated above, further information was sought by the regulator and more work was required at Day 70 of the MAA which the Defendant was unable to provide and undertake in view of the suspension of its manufacturing licence, in breach of the obligation contained in Clause 4.1 recited above;
(5) Mr Sinai contended that accepting the Defendant’s interpretation of clause 4.1 makes no business sense, because it means that the Claimant was paying for process validation batches and data which then could not be used in the MAA, if the Defendant decided to close its facility or relinquish its manufacturing licence. The Defendant’s interpretation is commercially unrealistic because it would mean that:
(i) The Claimant incurred very significant expenditure for creation of validation batches but took all the risk that such batches could not be used for the precise purpose for which they were created, simply because of circumstances brought about entirely by the Defendant;
(ii) The Defendant gave an express undertaking to manufacture validation batches in accordance with GMP, but its obligation had lapsed by the time that its validation batches and its processes came to be assessed by the regulator;
(6) Paragraph 9.1 of Annex 2 (and therefore a term of the DA) included stability testing (which is also expressly included in clause 3.1 on the Subject Matter of the DA). Testing was extended to 36 months by an additional scope of works on 1 November 2012. Stability testing (and therefore the Development) was not completed and had to be transferred to Recipharm when the Defendant closed its site at the end of April 2015;
(7) Dr Gamlen was not prepared to accept that the Development Plan in Annex 2 of the DA was “doomed to fail” as work required for a MAA. He said that Annex 2 was not of itself sufficient, but the issue would depend on what was actually included as the specific requirements of the process validation and the skill with which the process validation protocol is written;
(8) The Dossier was not limited to the IMPD. Mr Sinai relied upon the following:
(i) Dr Gamlen confirmed that process validation is not required for a standard sterilisation process and only for a generic MAA, that the clinical trials were in phase 2 for which process validation would not be required , and there was no reason that he could think of as to why an IMPD would have to be filed in CTD format ab initio ;
(ii) As stated at paragraph 68(4)(ii) above, the definition of “Dossier” expressly refers to an expert’s report: Dr Gamlen said that the reference to an expert’s report in the definition of Dossier is proof that the Dossier was for a MAA .
(9) The references in the DA to “quality control” and “regulatory approval” are not references to IMPD. Mr Sinai relied upon the following:
(i) the references to “registration” and “market authorisation” in the DA;
(ii) when looking at the wider context and factual matrix:
(a) the Defendant’s Detailed Development Plan P-258 refers to “licence submission” at paragraph 1.0 entitled “SCOPE” and at paragraph 6.0 to “Design Inputs: Quality Target Product Profile”. According to Dr Gamlen, these are only relevant to MAA ;
(b) the parties wanted the Defendant to become the commercial manufacturer of the licensed product and it is admitted that Ms Beveridge prepared the 5 year plan for commercial manufacturing. Mr Sinai accepted, however, that the 5 Year Plan was a hope which in the absence of a commercial manufacturing agreement is equally consistent with a fill and finish function. The highest he put it was that, when the DA was entered into a commercial manufacturing agreement was definitely “on the radar”;
(10) Finally, Mr Sinai relied on the parties’ subsequent conduct as evidence of their contractual intention as it existed when the DA was entered into. In particular he pointed to:
(i) A letter dated 9 May 2014 from Alan Krol, the Defendant’s Managing Director, which stated: “we are unable to inform you as to precisely how long we will cease manufacturing and the impact that this will have on our contract with you.”;
(ii) An email dated 14 August 2014 from Ms Krupa Gokani, a Project Manager of the Defendant to Dr Strieder, where the Defendant sent its replies to AGES’ Product questions for the MAA (without any further charges as this was included in the DA);
(iii) The agenda for the meeting of 30 September 2014 in London where one discussion item for the afternoon was “additional work required by Moorfields including request from Diapharm for QA/regulatory information”;
(iv) An email from Dr Strieder to Diapharm dated 1 October 2014 following the meeting in London where he wrote that it was decided that Diapharm’s answers would be responded to jointly by the Claimant and the Defendant by the end of November 2014;
(v) The draft Deed of Termination sent on May 2015 seeking to terminate the DA and to release the Defendant from all liability, which document is wholly inconsistent with the Defendant’s Defence herein and the assertion that its obligations were limited to “fill and finish” of the 12 validation batches already manufactured.
69. In closing Mr Sinai submitted that on the basis of Dr Gamlen’s evidence, the Court should have no difficulty implying that the requirement to maintain GMP until final determination of the MAA is both necessary for business efficacy and so obvious as to go without saying:
(i) If the GMP licence of the named manufacturer and site were suspended, then the process validation data was not going to be approved; Dr Gamlen stated: “the link between the process validation data and the physicality of the site is absent” ;
(ii) Dr Gamlen confirmed the opinion in paragraph 34 of his first report that “the requirement for the manufacturer to maintain their GMP status during, and after completion, of the development work is absolute” ;
(iii) When questioned by me about clause 4.1 of the DA, Dr Gamlen said that all the data which is submitted for a market authorisation application would have to be generated under GMP ;
(iv) It was Dr Gamlen’s opinion that it is standard to state in section 3.2.P.3 of the CTD that the named manufacturer would manufacture the Product in accordance with GMP ;
(v) Dr Gamlen confirmed the point made in paragraph 25(v) of the Amended Particulars of Claim that it is standard industry practice for the named manufacturer to continue to develop the Product until the Claimant’s MAA was finally determined .
70. Mr Lomas relied upon the fact that the DA is a detailed and professionally drafted agreement. Thus, the focus of the interpretive exercise should be to analyse the text used rather than to resort to extraneous matter. In this regard he relied upon the cases of Rainy Sky, Arnold v Britton and Wood v Capita referred to at paragraph 65 above. There is no ambiguity in the express terms which needs resolving.
71. On the Defendant’s case, the entire scope of the DEVELOPMENT must fall within the four corners of the Development Plan at Annex./2, as updated by mutually agreed Work Orders. He submitted that this is ultimately a matter of interpretation, in circumstances where there is no sole or predominant purpose ascribed to the batches produced by the Defendant under the DA, the definition of “DEVELOPMENT” under the DA merely requires “all work” to be done, i.e., batches to be manufactured (and related data to be generated) that was necessary to “fulfil the demands” of:
(1) the Guideline on the Requirements to the Chemical and Pharmaceutical Quality Documentation Concerning Investigational Medicinal Products in Clinical Trials (October 2006) by the EMEA CHMP; and
(2) the Notice to Applicants, Volume 2B, incorporating the Common Technical Document (CTD Part 3.2.P) (May 2008) by the European Commission except for chapter 3.2.P.2.
72. The extent of “all work” was clarified as being: (i) “the transfer of the production process from a former manufacturer”; and (ii) the manufacture and analysis of three batches. The scope of the Defendant’s obligations in respect of DEVELOPMENT is then defined by Clause 3.1 by reference to Annex./2. Annex./2 sets out nine discrete activities compromising the totality of the DEVELOPMENT, which he describes in detail at paragraph 128(b) of the Defendant’s opening written submissions. I do not reproduce them here. Suffice it to say, he submitted there is nothing in the DA that even mentions a MAA, let alone supports the contention that the Defendant was under an obligation to support one. The reference to an IMPD was indicative that the work concerned was at a far more embryonic stage of drug development, where clinical trials had not yet even been approved to start.
73. Manifestly, the three batches (and the related analytical data) did fulfil the demands of both documents: clinical trials were conducted, the data (including stability data) was used for the IMPD, and the Defendant was contracted under a separate work order (as an obligation outside the scope of the DA) to produce a first draft of Module 3 in reliance on the batches and the data, i.e. the data was manifestly considered by the Claimant as meeting the demands of the CTD and was able to be “plugged in” to the Notice to Applicants and the CTD.
74. To succeed the Claimant must either (i) establish that the DEVELOPMENT does not end until final determination of an MAA; or (ii) imply into clause 4.1 words that make retaining GMP status after DEVELOPMENT a free-standing obligation.
75. There is no basis on the face of the DA to read in an obligation on the part of the Defendant to maintain GMP until final determination of a generic MAA. Nor is there any commercial rationale for implying, as standard, a term to the same effect: industry practice was not followed in a key respect: the Claimant did not mitigate the risk of the Defendant not becoming manufacturer by entering into a supply agreement, so there is no basis to presume that the parties intended industry practice to apply in other areas. Thus, in the absence of an obligation, there can be no finding of breach.
76. It is important to note that in this context the definition of DEVELOPMENT is limited only to work for filing an IMPD - namely an application to approve using Investigational Medicinal Products in Clinical Trials. This is supported by: (i) the existence of and interpretation of the term DOSSIER at Clause 2.1, and the obligation at Clause 4.6 for the Defendant to hand this to the Claimant. DOSSIER is clearly a reference to an IMPD; and (ii) the fact that an IMPD often follows the same format of the CTD (also known as Module 3).
77. Specifically, there is no mention of an MAA or securing an MA at all: this is entirely rational as both of these are activities that would only take place after clinical trials had been successfully concluded. Further, the general definition of DEVELOPMENT in clause 2.1 is circumscribed by reference to the Defendant’s obligations to perform only certain DEVELOPMENT activities by reference to clause 3.1. Dr Lomas highlighted the particular words in that clause as follows:
“[the Defendant] shall perform the DEVELOPMENT in accordance with the directives given by [the Claimant] in writing and in accordance to the development plan given by [the Claimant] to [the Defendant] (Annex./2). The DEVELOPMENT comprises all necessary development steps for the manufacture and the chemical-pharmaceutical part including long-term stability and quality control. Additional subject matters have to be agreed on in writing and signed by both Parties. It is agreed and understood between the Parties that, contingent on the development character of this project, [the Defendant] does not assume any responsibility for the successful DEVELOPMENT of the PRODUCT and the regulatory approval of the product”.
78. An IMPD is a dossier comprising one of several pieces of IMP related data required in support of a clinical trial application, which is required ahead of the performance of a clinical trial in one or more EU member states. It is not a MAA, which can only be filled after completion of successful clinical trials, which often take many years to conclude.
79. Dr Lomas relied upon the evidence of Dr Gamlen that the work described in Annex./2 was insufficient in itself to support an MAA, and that this therefore suggests that was not the purpose of the DA. Mr Gamlen’s evidence at point 32 of his second Report dated 25 February 2021 was:
“Limitation of process validation to filtration and feasibility studies, as stated in Annex 2, would make the data incomplete and unsuitable for a marketing authorisation application. More work than is listed in Annex 2 would be needed to meet marketing authorisation application standards.”
80. He also relied upon the following response by Dr Gamlen to the Part 35 Questions from the Claimant:
“The Process Validation Protocol for a product for which a Marketing Authorisation Application should be reviewed by the Qualified Person intended to be responsible for product release once the product is approved. There is no mention of a Qualified Person in any of the documentation which I have been shown although the Qualified Person must have been named on the MIA held by the Defendants. This, combined with the deificiencies (sic) found by the assessor, makes me think that the protocol was in fact written for IMP manufacture and not MAA.
The contents of a Development Agreement clearly vary from company to company depending on the size of the Parties. In general large organisations will incorporate detailed control whereas smaller organisations will include less detail (often because they are relying on the contractor to know what is needed). If the Development Agreement is intended to generate a full data set for a market authorisation application then it should include the process for reviewing quality documents (such as the Process Development Protocol) and the process for generating , reviewing and approving the Marketing Authorisation Submission including a review by the QP (or in a large organisation the QP’s office) as they will be responsible for the release of the product onto the market. None of these things were included in the schedule of work provided by the Plaintiff to the Defendant. For this reason I do not think that Schedule 2 was written to include generation of data to support an MAA.”
81. The provision in clause 3.3 of the DA which provides “CMO shall perform the DEVELOPMENT of the PRODUCT exclusively for COM.”, effectively binds the Defendant not to manufacture a competing version of the Product for a third party. It is not, as the Claimant suggests, an obligation that the Claimant will work only with the Defendant on the Product.
82. When the Defendant lost its GMP licence and was preparing its Risk Assessment in mid‑2014 to assess the impact of transferring to an alternative manufacturer, no mention of MAA was made. Instead, the regulatory requirements were described as being that:
“[the Claimant] would manage the update and submission for the IMPD and the clinical trial protocol.”
83. There were four further points relied upon by Dr Lomas:
(1) The Work Orders point - Clause 3.1 stipulated that “additional subject matters have to be agreed in writing and signed by both parties.” This led to a number of work orders (“Work Orders”). Those are out at Annex 1 of the Defendant’s written submissions and covered areas such as the manufacture and release of additional small batches of the Product for compassionate use and what he described as “fairly trivial/small work streams” such as packaging and labelling, storage and distribution; analytical testing and stopper compatibility study - including testing. All of these supported the Defendant’s contention that the scope of the DA was narrow and limited to certain prescribed manufacturing obligations towards manufacturing material that might be used for clinical trials. Indeed, if the DA was intended to support a MAA (as contended for by the Claimant), one would expect to see Work Orders for more than labelling or studying the stoppers of the vials being used for three process validation batches - essentially work to show that the manufacturing process was reliable and consistent in output;
(2) If the Claimant wanted additional services, such as the manufacture of additional batches (or the preparation of the Module 3 documentation ), then it was necessary for the parties to agree such additional services under new Work Orders, including an adjustment to the price. Until such time as a Work Order was executed (and there is no obligation compelling either party to issue or accept new Work Orders), there were no future obligations on either party. Either party was free at any time to say “no”;
(3) The disjunctive “and” point - As stated at paragraph 23(3) above, the last sentence of clause 3.1 provided:
“It is agreed and understood between the Parties that, contingent on the development character of this project, [the Defendant] does not assume any responsibility for the successful DEVELOPMENT of the PRODUCT and the regulatory approval of the product”.
The highlighted “and” in clause 3.1 is disjunctive. It serves to differentiate as between DEVELOPMENT on one hand (that the Defendant had to carry out under GMP) and regulatory approval (i.e., a MAA) on the other, i.e., supporting a MAA does not fall within the scope of the DEVELOPMENT. Support for this also comes from the fact that the Module 3 documentation (the CTD, which is also part of an IMPD) was completed by the Defendant under an additional scope of Works Order , i.e., even a document as central on the Claimant’s case to a MAA was not within the remit of the DA as drafted;
(4) The “no responsibility point” - Clause 3.1 confirms that the Defendant does not assume responsibility for the successful DEVELOPMENT of the Product and regulatory approval of the product. This contradicts any suggestion that the Defendant was under a long term obligation to “wait in the wings” and support the Claimant in its development of the product. The Defendant’s obligation was to undertake, which it did, the work in Annex 2 and the Work Orders; anything else would be subject to the parties reaching some future agreement(s) which each had the discretion to accept or reject.
84. This can be stated shortly. The Defendant’s position is that in order to imply what Dr Lomas describes as “an evergreen GMP obligation” for the Claimant’s benefit, one would have to establish a standard industry practice. Specifically, the Court will only imply terms to give business efficacy to a contract or because the words to be implied are so obvious as to go without saying. In the instant case, it is unclear how the Claimant can possibly satisfy either test in the context of: (i) a comprehensive and professionally drafted agreement; and (ii) expert evidence that suggests that, to the contrary, it would not be obvious to import an obligation onto the Defendant in the absence of a further Supply Agreement.
85. Whilst I entirely accept that no supply agreement was concluded between the parties, part of the relevant factual matrix was that at the time the DA was executed, the parties were operating on the basis that a supply agreement would be entered into, with the Defendant named as the commercial manufacturer. Had the Defendant’s GMP licence not been taken away, the intention of the parties was that after approval, the Defendant would have gone on to become the manufacturer and have entered into the Supply Agreement. One is entitled to take this into account when determining what the true ambit of the obligations were under the Development Agreement.
86. Applying the relevant principles of contractual construction set out at paragraph 65 above, I would draw attention to the following:
(1) Under clause 4.5, the Defendant committed to co-operate with the Claimant to achieve “Clinical Study Approval and Marketing Authorisation for the Product.”;
(2) In his oral closing submissions, Dr Lomas submitted that a key question was what was meant by the words “all work” in the definition of DEVELOPMENT in clause 2.1 of the DA. It is to be noted that an integral part of the work expressly included the taking of “all measures to ensure a smooth production of three validation batches - including but not limited to filtration studies, stress tests, analytical method transfer.”
87. In my judgment that work and the Defendant’s obligations under the DA were not limited to a simple “fill and finish” agreement to produce 12 clinical study batches which it produced. They went further than that. I reach that conclusion for the reasons set out below.
88. Looking at the objective meaning of the language which the parties have chosen to express their agreement, the Defendant’s obligations under the DA related both to the manufacture of the drug for clinical trials, providing data to create the IMP and the IMPD and the production of validation batches of the drug for the purposes of the MAA. Paragraph 20(a)(i) of the Defence accepts that the production of the validation batches was to be used for both purposes.
89. The Defendant’s interpretation of clause 4.1 restricting its obligations to the production of the 12 clinical study batches is, in my view, commercially unrealistic, because it means that those batches and the related data could not be used in relation to the MAA, if the Defendant lost its manufacturing licence or closed down its manufacturing facility. It is also inconsistent with clause 4.4 of the DA, which I address at paragraph 94 below.
90. In my judgment the DOSSIER does not only relate to the IMPD. The definition of DOSSIER in clause 2.1 includes reference to “an expert’s report” that is only consistent with work in relation to a MAA. Dr Gamlen stated that: “All of the documentation that… I have been shown confirms that this was, indeed, preparation for market authorisation.” . Clause 4.1 required the Defendant to perform all of DEVELOPMENT in accordance with the GMP. In my view that was a continuing obligation.
91. Clause 4.4. contained an obligation on the part of the Defendant to complete the DEVELOPMENT “in accordance with the development plan Annex./2 of the DA”. Annex./2 of the DA contained provisions which are only consistent with a continuing GMP licence being in place after production of the 12 clinical study batches produced by the Defendant. For example, it expressly referred to “the Stability Studies Finished Product” and within the “Illustrative Development Timeline” expressly referred to “Module 3 documentation”, which is only concerned with commercial manufacturing and is thus required for the MAA. Paragraph 20(1) of the Defence admits that “the preparation of Module 3 documentation fell within the scope of the DEVELOPMENT, as evidenced by the timeline at Annex./2, the preparation of Module 3 documentation was not the exclusive responsibility of the Defendant since the majority of the Module 3 requires information beyond the records and work contracted to be undertaken by the Defendant.” The definition of DEVELOPMENT excludes only chapter 3.2.P.2. of Module 3. On 11 March 2013, there was an Additional Scope of Work order, entitled “Generation of Module 3”, which covered some drafting by the Defendant and for which it was paid a modest sum of £6,573. It appears that this work was to have been done by or on behalf of the Claimant and instead the Defendant agreed to it. In my judgment, this does not detract from the fact the Defendant had already performed process validation for the MAA pursuant to its obligations under the DA and this work was lost when its GMP licence was suspended.
92. Clause 4.5 of the DA contained a continuing obligation on the part of the Defendant to provide information relating to the DEVELOPMENT and the PRODUCT to any government authority at its request within 15 days at no further cost. That required the GMP licence to be in place. As Dr Gamlen stated, whilst it was standard industry practice to have a manufacturing agreement in place, the fact that there was not in the present case, does not prevent a MAA being pursued.
93. Annex./2 also provided that “Timelines will be agreed between the parties following signature of contract by both parties.” Those timelines were contained in the Detailed Development Plan, which I have found at paragraph 23(11) above was created after the DA was entered into, and in my view, given its being envisaged within the DA itself, is something to which I am entitled to have regard when construing the true meaning of the DA.
94. I do not accept Dr Lomas’ submission that the process validation exercise related to the IMPD and clinical trials. In this regard I accept the evidence of Dr Gamlen that the heading “6.0 Design Inputs: Quality Target Product Profile” in the Detailed Development Plan is “a specific regulatory term which refer to the development of generic products for marketing authorisations” , and the reference to “Regulatory and statutory requirements of the intended markets” within that heading has to be read in that context.
95. I accept Mr Sinai’s submission that the Defendant was contracted to produce process validation, which was in accordance with GMP, and which could be used in its MAA. When the Defendant lost its GMP licence, the process validation that it created could no longer be used in the MAA and had to be redone.
96. Dr Gamlen was not prepared to accept the point put to him by Dr Lomas that the Development Plan set out in Annex./2 was doomed to failure as work required for a MAA. He said that the descriptions in Annex 2 “were not of themselves sufficient to completely define an MAA…”, but much would depend on the content of specific requirements of the process validation and the skill with which the process validation protocol was written. The Detailed Development Plan contained more detail and included at 6.0 “Design Inputs: Quality Target Product Profile”, which, as stated at paragraph 97 above, Dr Gamlen said this heading is a reference “to the development of generic products for marketing authorisations.”
97. Further, in any event, it seems to me that whether or not the Development Plan would result in a successful outcome, it does not impact on the issue on whether the Defendant’s obligations under the DA related both to the manufacture of the drug for clinical trials, providing data to create the IMP and the IMPD and the production of validation batches of the drug for the purposes of the MAA. As paragraph 20(v) of the Particulars of Claim accepts that obligation contained no promise that that Product could be successfully manufactured or that a MAL would be obtained.
98. A new point was taken in submissions by Dr Lomas, namely that the process validation was for an unrelated MAA for CTREPH, to which I have referred at paragraph 54 above. This Defence has not been pleaded and no application was made to amend so as to add it. The amended Particulars of Claim is pleaded on the basis of a generic MAA. Specifically paragraphs 32 and 33 therein, plead Diapharm’s request to the Defendant for GMP declarations. Those facts are admitted at paragraphs 26 of the Defence, but their relevance is denied. The Defendant did not there advance a defence that the DA was not intended to cover a generic MAA.
99. In the light of the findings on the construction of express terms of the DA, in my judgment it is not necessary to imply any of the terms contended for by the Claimant. I accept the Defendant’s submission that where one has a professionally drawn agreement, one should be reluctant to import implied terms, unless it is necessary to give business efficacy to the DA or because the words to be implied are so obvious as to go without saying. Neither is the case here.
100. I turn to consider each of the issues contained in the List of Issues approved by the Court.
101. Did the Defendant’s obligations under the DA require it to manufacture validation batches to be used as part of the Claimant’s MAA application, or was the Defendant’s obligation limited to a fill and finish function?
102. As stated above, I find that the Defendant’s obligation was not limited to a fill and finish function but required it to manufacture validation batches to be used as part of the Claimant’s MAA.
103. Did the Defendant’s obligations under the DA require it to be named as the commercial manufacturer in the Claimant’s market authorisation application? If yes, on what terms did that obligation arise?
104. The process validation information is intended to demonstrate that the proposed manufacturer named in the application form can consistently reproduce the drug in question under the same manufacturing conditions. Validation is specific to the designated manufacturing site, i.e., the validation data produced and submitted by the Defendant could only be used to validate the Defendant’s manufacturing site and processes.
105. Given that the definition of Development included “all work necessary to fulfil the demands of the… CTD” and clause 4.5 contained an undertaking to co-operate to “achieve Marketing Authorisation” and to use the Defendant’s best efforts to support further activities required by the market regulator, taken together with the fact that at the time the DA was entered into the parties were operating on the basis that a supply agreement would be entered into with the Defendant named as the commercial manufacturer, it is inconceivable that anyone other than the Defendant would be named as the commercial manufacturer in the MAA.
106. Indeed, the Defendant itself recognised that it was required to be named as the commercial manufacturer in the MAA. In particular:
(1) it named itself as the “Drug Product Manufacturer” in Section 3.2.P.3.1 of Module 3 that it prepared;
(2) thereafter in November 2013 Section 3.2.P.3.1 was filed in the MAA with the Defendant named as the proposed manufacturer;
(3) when AGES later sought clarification about the identity of the finished product manufacturer, the Defendant’s response to AGES’ question 33 was that it should be named as the manufacturer of the finished product.
It is difficult to see which entity at that stage could have been identified as the commercial manufacturer at the time the MAA was prepared by the Defendant and filed.
107. Did the Defendant’s obligations under the DA require it to maintain its European current GMP accreditation throughout the Development and until final determination of the Claimant’s market authorisation application?
108. Yes, it did, for the reasons set out above.
109. If the answer to 1(iii) is yes, was there any date on which this obligation would cease?
110. Dr Gamlen’s evidence, which I accept, was that when performing clause 4.5 of the DA, it is standard industry practice for the named manufacturer to continue to develop the drug until the Claimant’s MAA was finally determined. As referred to at paragraph 97 above, as the Claimant accepts at paragraph 25(v) of the Particulars of Claim, that obligation contained no promise that that Product could be successfully manufactured or that a MAL would be obtained.
111. Did the Defendant’s obligations under the DA require it to carry out stability testing on the validation batches it manufactured at regular intervals for up to 36 months?
112. Yes, it did, pursuant to the definition of ‘DEVELOPMENT’ in clause 2.1 and clauses 4.1 and 4.4. of the DA.
113. Did the Defendant’s obligations under the DA require it to manufacture four clinical trial batches for release by mid-October 2014?
114. Yes, it did. Under the definition of ‘DEVELOPMENT’ in clause 2.1 and clauses 4.1 and 4.4 of the DA, the Defendant’s obligations included carrying out all work required for the demands of the clinical trials. The immediate voluntary suspension in manufacturing on 19 December 2013 arose because of MHRA’s findings of critical failures by the Defendant to comply with certain aspects of GMP. The Defendant was clearly aware of the effect that suspension would have on stock supplies, hence the letter dated 9 May 2014 from Alan Krol, the Defendant’s Managing Director, which stated that the Defendant was not permitted to re-start manufacturing activities, pending a MHRA inspection scheduled for 14 May, expressing (over-optimistically as it transpired) that “We are confident that we will be able to reassure the MHRA and return to business as usual shortly.” That did not happen. On 29 May 2014, Krupa Gokani, a project manager at the Defendant wrote to Dr Strohmaier, the Claimant’s Managing Director and Ms Tan, stating “As confirmed by yourself, we would expect to require another full production run (4 batches, I for each strength) in late August / early September for release by mid‑October.”, and informing them that it would put together a case to the MHRA “to justify the continued production at Moorfields”, on the basis that there would not be sufficient stock to continue the clinical trial if the process was transferred elsewhere. This was the clearest acknowledgement of such an obligation. The attempt to persuade the MHRA was unsuccessful and in the event the only course of action available was to obtain the manufacture by Recipharm of one emergency batch of 10 mg strength because there was a critical need for at least that amount, on the basis that the Defendant would release, label and package the batch.
115. Did the Defendant complete the Development?
116. No, for the reasons stated above. In the absence of a continuing GMP licence, the Defendant did not complete the stability testing it was required to do; due to the cessation of manufacturing, it did not complete all the batches, and further it was unable to complete the work required by the demands of the CTD.
117. Did the Claimant file an application for market authorisation?
118. Yes. See paragraph 37 above. The application was filed by Diapharm, acting as agents for the Claimant, as Dr Risse, Diapharm’s Senior Manager, Regulatory Affairs, informed Ms Beveridge in an email dated 10 September 2014. The fact that Diapharm was acting as the agent of the Claimant in submitting the MAA to AGES was addressed in paragraph 15 of the second witness statement of Mr Beckers dated 21 February 2021, paragraph 3 of Dr Strieder’s witness statement dated 22 January 2021 and paragraph 3 of Ms Schuller’s witness statement dated 22 February 2021. I accept their evidence. Mr Beckers was the only one of those witnesses who was cross-examined by Dr Lomas about Diapharm making the MAA. The cross-examination appears at Day 2, p57, lines 7 to 26. There was no challenge to the explanation given there by Mr Beckers about the agency relationship. Dr Lomas simply said “Understood”.
119. Did the Defendant breach the Development Agreement by losing its GMP status by November 2014 and by closing its facility on January 2015?
120. Yes, for the reasons stated above. Their obligations under the DA were not completed. There was work outstanding pursuant to the Day 70 Report and the responses to many of the questions posed therein, which the Defendant was unable to carry out, notwithstanding their obligations under clauses 4.1 and 4.5 of the DA, not least because it had decided to close its facility, rather than seek to comply with GMP and reinstate its manufacturing licence.
121. Did the following matters occur and were they caused by the Defendant’s breach: Were the Defendant’s validation batches and data incapable of being used in the market authorisation application? Was the Defendant identified in the market authorisation application as the finished product manufacturer? Was the statement of non-compliance with GMP in the AGES Day 70 Report a determinative reason for refusing to approve the Drug? Was it necessary for the Claimant to submit validation batches from another manufacturer for the market authorisation application?
122. I take each of those points in turn.
123. Were the Defendant’s validation batches and data incapable of being used in the market authorisation application?
124. The Defendant’s validation batches and data, were to the knowledge of the Defendant, used in the MAA as part of the Module 3 submission. That formed part of its obligations under the clauses 4.1, 4.4 and 4.5 of the DA. Once, however, the Defendant’s activities were suspended for breaches of GMP, they could not be used in the market authorisation process. In the Day 70 Report, AGES stated that the MHRA’s statement of the Defendant’s non-compliance with GMP was “considered as a major objection”. Further work had to be done, which the Defendant was unable to carry out, and it could not make the changes which AGES required. These were breaches of the DA.
125. Was the Defendant identified in the market authorisation application as the finished product manufacturer?
126. Yes - see the points made at paragraphs 104 to 106 above.
127. Was the statement of non-compliance with GMP in the AGES Day 70 Report a determinative reason for refusing to approve the Drug?
128. Yes. Whilst I accept that there were a number of objections to the Product identified by AGES which required rectification, the application was bound to fail as a result of GMP failures at the Defendant.
129. Was it necessary for the Claimant to submit validation batches from another manufacturer for the market authorisation application?
130. Yes, because in breach of its obligations under the DA, the Defendant was unable to complete the work required. See paragraph 124 above.
131. Did the following matters occur and were they caused by the Defendant’s breach: was the Module 3 dossier prepared by the Defendant capable of being used in the market authorisation application?
132. The Module 3 dossier was prepared by the Defendant pursuant to its obligations under the DA and the additional work agreed on 13 March 2013, referred to at paragraph 33 above. It was capable of being used and was used in the MAA, but once the Defendant suspended manufacturing in December 2013 and was unable to carry out aseptic filling and sterile terminalisation in accordance with GMP, their validation process and data could not no longer be used. As stated at paragraph 124 above, further work had to be done, which the Defendant was unable to carry out, and it could not make the changes which AGES required. These were breaches of the DA, because the Defendant decided to close its manufacturing facility, rather than to comply with GMP and seek to reinstate its manufacturing licence.
133. Was it necessary for the Claimant to arrange for the Module 3 submission to be redone?
134. Yes, by reason of the Defendant’s breaches of the DA referred to above, and their inability to being able to carry out any remedial work to address the issues identified by AGES, it was necessary for the Claimant to arrange for a Module 3 submission to be redone.
135. Did the following matters occur and were they caused by the Defendant’s breach: was the Defendant unable to respond to the regulator’s questions and requests for information, and to make any changes to the Drug determined necessary by the regulator as part of the application process.
136. Yes - see paragraph 135 above.
137. Did the following matters occur and were they caused by the Defendant’s breach: did the Defendant fail to manufacture clinical trial batches for release by October 2014 and did the Claimant have to manufacture an emergency batch through Recipharm instead?
138. Yes, in all respects. See paragraphs 42-45, 51-54 and 131 above.
139. Did the following matters occur and were they caused by the Defendant’s breach: did the Defendant fail to complete the stability testing making it necessary for the Claimant to instruct Recipharm to complete the stability testing, or did the Claimant consent to Recipharm carrying out the stability testing irrespective of the Defendant’s breach?
140. In breach of the DA, the Defendant failed to complete the stability testing making it necessary for the Claimant to instruct Recipharm to complete the stability testing. This was a decision imposed upon them by the Defendant’s inability to carry out such work.
141. In this regard, I would refer to the telephone conference call which took place on 3 February 2015, following closure of the Defendant’s site, referred to at paragraph 56 above at which Dr Strohmaier, Dr Strieder and Ms Tan were informed that the Defendant would cease all activities around the end of April 2015, including manufacturing, packaging and stability. As a result, the Claimant could no longer complete the stability testing on manufactured batches and it was required to transfer stability and packaging to another manufacturer, Recipharm.
142. Was it necessary for the Claimant to purchase replacement API in order to submit new validation batches from Recipharm for the market authorisation application? If so, did the Claimant mitigate this loss when using and administering the replacement API?
143. I accept the evidence of Dr Gamlen that it was necessary for the Claimant to purchase replacement API in order to submit new validation batches for the MAA, because the Defendant had lost its GMP licence.
144. Has the Claimant incurred the losses pleaded in paragraph 55 of the Particulars of Claim? Are any losses incurred recoverable? Has there been a failure to mitigate any losses?
The sums claimed by the Claimant
145. Under paragraph 55 of the Amended Particulars of Claim, the Claimant claims the sum of €1,794,932. The loss is pleaded under five separate headings:
(A) Production and Validation
|
(i) Technical transfer from Moorfields: |
€66,645 |
|
(ii) Cost of replacement API: |
€1,162,786 |
|
(iii) Testing of API: |
€21,178 |
|
(iv) Credit for API larger batch sizes: |
-€354,104 |
|
(v) Production and documentation: |
€453,515 |
|
(vi) Validation: |
€82,800 |
|
Sub Total: Production and Validation: |
€1,432,820 |
(B) Stability Study and Module 3
|
(i) Stability Study: |
€68,600 |
|
(ii) Preparation of Module 3: |
€12,000 |
|
Sub Total: Stability Study and Module 3: |
€80,600 |
(C) Finalisation Stability Study
|
(i) Stability Study Finalisation: |
€22,500 |
|
(ii) Delivery of samples to ReciPharm: |
€6,501 |
|
Sub Total: Finalisation Stability Study: |
€29,001 |
(D) Emergency Drug Batch
|
(i) Production Emergency Batch: |
€59,056 |
|
(ii) Cost for analysis and release: |
€2,048 |
|
(iii) Auditing ReciPharm: |
€5,515 |
|
(iv) Transfer costs API and Batch: |
€7,965 |
|
(v) Credit for standard production costs (that Moorfields would have charged): |
-€6,873 |
|
Sub Total: Emergency Drug Batch: |
€67,711 |
(E) Claimant’s Additional Expenses
(a) Generic Submission
|
(i) Evaluation of CMO’s/Tech Transfer/ Supervising/Production set up: |
€134,400 |
|
(ii) Prolongation of clock stop/Development and Module 3/Re-submission: |
€28,800 |
(b) Clinical Study
|
(i) Negotiations for continuing use of Clinical Trial Material from Moorfields: |
€9,600 |
|
(ii) Patients’ safety; vial substitution/ relabeling/discontinuation of recruitment |
€12,000 |
|
Sub Total: Additional Expenses |
€184,800 |
|
Total loss: |
€1,794,932 |
146. These figures are supported by an amended Schedule of Loss which contains a breakdown of the amounts claimed by reference to specific invoices and working hours. In relation to the figure of €1,162,786, relating to the cost of replacement API claimed (see paragraph 145(A)(ii) above), there is additionally an Annex A, which contains a further breakdown of the relevant invoices, which were charged in US dollars, showing the US$/€ conversion rate at the material date. Ms Tan at paragraph 34 of her witness statement dated 22 February 2021 as amended, stated that there had been an error in Annex A and amended the figure claimed for wasted API, which had been over-calculated by 3.7g in relation to full sample testing. This result in a reduction of €18,087.73.
147. At the hearing I indicated to Counsel that, having determined liability, I would then address the principles which would be calculated, and I would then hear further argument from the parties, in the absence of agreement on quantum . I will set out first the Defendant’s submissions relating to this claim and then the Claimant’s response to the points made. There are a number of aspects in relation to the disputed figures on which I will need to hear further submissions, in the absence of agreement.
148. The Defendant submits that the Claimant has failed to establish any losses caused by the breaches of the DA, alternatively that the Claimant has failed to establish that it had incurred any losses.
149. The essence of Defendant’s case is that:
(1) there was no obligation on the Defendant to enter into a Supply Agreement;
(2) it is common ground that any change in manufacturer would require de novo process validation. Both Ms Schuller and Ms Tan accepted that a new manufacturer would have meant redoing the process validation;
(3) most of the costs sought to be recovered were in fact expenses incurred by CompLex, a third party, with no evidence of recharging to the Claimant, no cost sharing agreement with the Claimant and no evidence of any controlling relationship between the Claimant and the party which incurred the costs.
150. In the further alternative, in relation to mitigation, the Claimant ordered significantly larger batch sizes when manufacturing with Recipharm, and, in its claim, has given the Defendant a credit in this respect. However, according to the evidence of Ms Tan, there were 72.63 g of API (amounting to US$479,358) for which there was no purpose to which it could be put, and it was destroyed. It is not understood why the Claimant ordered so much more API and was then left unable to use it. In any event, the Defendant should not be liable for the Claimant’s inability to plan properly.
151. In relation to the Claimant’s Annex A, there is no real clarity as to why some (and not all) of the API on certain invoices is claimed, or how apportionment between the sums sought under this claim and another (unidentified) treprostinil project was undertaken. Similarly, the basis for apportioning the testing amounts in respect of each invoice is not comprehensible as these do not follow the same proportions of invoiced / claimed. For example, in relation to two of the invoices in Annex A:
(1) CG-1030926: 15.8 g (21%) of total invoice amount is claimed, but within that 15.8 g, 8.8 g (56%) was used for testing; and
(2) CG-104050403 7.36 g (14.6%) of total invoice amount is claimed, but within that 5.1 g (69%) was used for testing.
152. The Claimant has now put forward a fourth iteration of loss. Ms Tan has admitted that earlier errors were made in her previous calculations. Furthermore, the process by which the Claimant allocated losses within invoices for larger sums is unclear. These aspects and the matters referred to at paragraph 151 above, put into doubt any sense of accuracy.
153. €993,482 (once credit is given for larger batch sizes) of the losses claimed were incurred by CompLex, not the Claimant. First, all the relevant invoices were between the manufacturer and CompLex. The evidence of Mr Beckers was that the Claimant was not part of the same group as CompLex at the material time. There is no written agreement in relation to any repayment of costs incurred by CompLex, and no evidence has been produced by the Claimant to show that it has paid for any of the replacement API. There is no contractual mechanism in place between them for repayment. Additionally, the figure of €184,000 relating to “additional expenses” includes work done by Ms Tan and Ms Schuller, neither of whom were employed by the Claimant at the material time. Again, there is no evidence of any obligation on the part of the Claimant to reimburse their employers, CompLex in the case of Ms Tan, and Amomed in the case of Ms Schuller, in respect of this work.
154. The Claimant’s reliance on the Court of Appeal and Supreme Court decisions in Swynson Ltd v Lowick Rose LLP, [2015] EWCA Civ 629, [2017] UKSC 32 in support of recovery of the Chirogate invoices paid by CompLex is misplaced. The key difference between this case and Swynson is that the claimant in Swynson had incurred the loss (in respect of a loan), but this loss had then been extinguished (by a further subsequent loan). Here there is no loss to the Claimant: it has not incurred the costs or paid the sums sought at §55(A)(ii) and (E) the amended Particulars of Claim and is under no obligation to pay CompLex. Both Lord Sumption at [11] and Lord Neuberger at [98] indicated that the House of Lords decision of Parry v Cleaver was not to be regarded as authority for “deciding each case in what may be regarded as its broader commercial merits” or “as a green light for doing whatever seems fair on the facts of the particular case.”
155. The Claimant has therefore not lost the sum of €993.482 claimed. If one deducts the figure of €993,482 from the sum of €1,794,932, that leaves a maximum figure recoverable of €801,450.
156. Further given that:
(1) the evidence of Mr Strieder was that the Defendant was not under an obligation under the DA to produce a Module 3 draft , and
(2) the evidence of Ms Tan was the Defendant was not under an obligation under the DA to produce additional clinical supply batches
the claim for Stability Study and Module 3 (paragraph 55(B) of the amended Particulars of Claim) and Emergency Batches, which were required for ongoing clinical study participants (paragraph 55(D) of the amended Particulars of Claim) also fail. These amount to €148,311, reducing the maximum figure recoverable to €653,139.
157. Of this remaining amount of €653,139:
(1) Technical transfer from Moorfields (paragraph 55(A)(i) of the amended Particulars of Claim) would have been incurred in any event , as the Claimant accepted the Defendant was not obliged to manufacture future batches. Therefore, a further €66,645 should be deducted;
(2) The testing of API would have been incurred in any event as more API always had to be purchased, alternatively would have been incurred from 1 January 2016 in any event. Therefore one should deduct a further €21,178 (alternatively, at a minimum deduct €8,773);
(3) Production and documentation (paragraph 55(A)(v) of the amended Particulars of Claim) again correlates to API being purchased (and should be either removed entirely or, in the alternative, adjusted to reflect and exclude the percentage of API bought after 1 January 2016). This is because, by reference to the Schedule of Loss, this breaks down into:
(i) Batch manufacture, €397,335. If the Claimant was buying API, it would have to manufacture it into a usable form. Dr Lomas referred to Ms Tan’s evidence on Day 3, p.41, lines 5-26, which to the effect that such costs would have been different depending on the number of batches being manufactured, it being cheaper to manufacture one big batch than twelve batches for product validation. The amount of such difference was not in evidence;
(ii) Analytical method validation costs of €33,500. Any new manufacturer would need to validate its analytical methods in any event;
(iii) Batch release analysis costs of €22,680. If more API was being purchased and manufactured into batches, these batches would need analysis on release. Therefore, one should deduct a further €453,515 (alternatively, at a minimum deduct €185,941 to reflect and exclude spending from 1 January 2016).
(4) Validation (paragraph 55(A)(vi) of the amended Particulars of Claim) as a head of loss is hard to assess. In light of the Claimant’s witnesses conceding that some validation would have to be done if a new manufacturer was used (or where there was a change in batch size / process), it is unclear how much of the €82,800 claimed would have been incurred in any event. By reference to the Schedule of Loss, this breaks down to:
(i) Invoice 640593: process validation consultant, €23,400;
(ii) Invoice 6600164: process validation, €19800.- (4 batches) / €9,900.- (2 batches);
(iii) Invoice 6600381: process validation for 6 batches €29,700.
(5) It is to be noted that six batches manufactured by Recipharm were contaminated with metacresol , so at least €29,700 should be deducted (if not the full amount given that validation would have to be redone to some extent anyway with batches not so contaminated);
(6) Finalisation Stability Study (paragraph 55(C) of the amended Particulars of Claim) is again hard to assess. It is not understood how these stability studies are different to those under the Module 3 Report. One therefore should deduct €29,001;
(7) As a general point Recipharm appear to be far more expensive for equivalent work (see, for example, the emergency batches which cost 10 x more than the proposal from the Defendant).
158. The Claimant accepts that the losses claimed must fall within the four corners of the DA. It was entitled to receive process validation which could be used for a MAA until it was determined. The Defendant’s obligation under the DA was to bring the drug to the point where a licence decision was made, and a commercial manufacturing licence hopefully obtained. Just as the Defendant has been at pains to state in these proceedings that it was entitled to walk away thereafter and to refuse to commercially manufacture the licensed drug, the Claimant was entitled to decide that it did not want to sell the drug, not appoint another commercial manufacturer and otherwise deal with the licensed product as an asset in its own right; or perhaps simply keep the licensed know‑how and approved licence / process as an asset.
159. In relation to the Defendant’s submission that the claimed losses would have been incurred on a change of manufacturer, given that there was no manufacturing agreement in place, Ms Sinai submitted that the losses claimed were incurred by reason of the Defendant’s non-performance of the DA. Matters which may or may not have happened post-development and post-breach, such as commercial manufacturing agreement or the identity of the entity that would be named as the market authorisation holder, do not, provide a defence to the Defendant in respect of loss caused by breach of the DA.
160. Reliance is placed by Mr Sinai on:
(1) the evidence of Ms Tan as to the amount of API required and the use, which was made of the API in sample testing, manufacturing batches of different strengths and administering what was possible to clinical trial patients. She also addresses why larger batch sizes needed to be manufactured by Recipharm in view of the fact that the development had moved on from November 2013 when the Defendant manufactured the initial strengths. Mr Sinai submits it was foreseeable that the requirements of the Development would change over time, that there was no obligation to manufacture the same size batches and the Claimant was entitled to produce volumes which represented the needs of the Development. Her spreadsheet sets out how 178.5 grams were used to create the 12 validation batches from Recipharm, how much of each of the batches of each concentration was used and for what, and that 68.1 grams was administered to patients in the clinical trials and 72.63 grams was subsequently destroyed because some of the batches expired as there was no use for them in the clinical trials;
(2) Recipharm’s Validation Report which lists the validation batches it manufactured by Recipharm by concentration and batch number;
(3) Annex A which sets out the weight of API purchased for each of those batches and traces the API in each batch to the relevant Chirogate invoice. It also sets out how much API sample testing needed to be carried out for each of those batches with reference to independent evidence from Recipharm setting out its testing Specifications.
These submissions do not, however, fully address the points made on behalf of the Defendant at paragraph 157 above.
161. Mr Sinai submitted that the issue comes down to whether the assistance provided by CompLex is avoided loss such as mitigation, in which case the Claimant cannot claim it, or whether it is a collateral benefit, or in old legal language, res inter alios acta, in which case it is to be disregarded.
162. The Claimant relied upon the decision of the Supreme Court in Swynson Ltd v Lowick Rose LLP [2017] UKSC 32. The case concerned a claim for damages for negligent accounting advice. The claimant, a lending company, made three loans to a borrower (“EMSL”) relying on a due diligence report prepared by the defendant, a firm of accountants. After EMSL defaulted on repayment of the loans, the claimant’s owner, Mr Hunt, personally loaned EMSL circa £16m under a loan agreement containing a condition that his loan be used to repay the claimant’s loan to EMSL. Mr Hunt caused the claimant’s loan to be repaid in this manner for tax reasons, and because he did not want a large unpaid loan to appear in the claimant’s books.
163. Both the judge at first instance, Rose J (as she then was) [2014] EWHC 2085 and the majority of the Court of Appeal (Longmore and Sales LJJ (as he then was), Davis LJ dissenting) [2015] EWCA Civ 629, held that Mr Hunt’s loan to EMSL which was then used to repay the claimant was collateral and thus the claimant’s claimed loss against the defendant was not avoided.
164. Longmore LJ at [10] stated:
“It is, of course, the law that an innocent party, who claims for breach of contract, is under a duty to take reasonable steps to mitigate his loss. In so doing, he may bring about a situation in which his loss is partly or wholly avoided. In this category of cases a question will arise whether that avoided loss has to be brought into account in assessing his damages. It may also be the case that a claimant's loss is partly or wholly avoided despite his taking no steps to mitigate his damages. The principles governing the assessment of damages are (or, at any rate, should be) similar in both categories of case. It is usually said that, if the transaction giving rise to the avoided loss arises by virtue of circumstances which are collateral to the breach of contract, the avoided loss need not be brought into account; but if the transaction giving rise to the avoided loss arises out of the consequences of the breach and in the ordinary course of business it is to be taken into account.”
165. Sales LJ, agreeing, stated at [53]:
“I consider, on the authority of Parry v Cleaver [1970] AC 1, that the principles governing whether some matter which reduces loss is to be regarded as collateral (or, in old legal language, res inter alios acta), and hence to be left out of account when deciding whether damages are payable in respect of that loss, are intended to reflect practical reality and basic justice as between the three persons involved: the person who has suffered the loss, the person who is in law responsible for causing the loss and the third party who has made a payment which reduces that loss. As Lord Reid said, at p 13H, “The common law has treated this matter as one depending on justice, reasonableness and public policy.” He went on to observe at p 15E, “the distinction between receipts which must be brought into account and those which must not must depend not on their source but on their intrinsic nature.”
166. The Court of Appeal upheld Rose J’s judgment on the basis that Mr Hunt’s loan was not mitigation since it was not brought about by the claimant, and whilst it had arisen because of the defendant’s breach, it had not arisen in the ordinary course of business and therefore it was a collateral matter which did not go to reduce the damages recoverable by the claimant from the defendant.
167. The Supreme Court overruled the Court of Appeal’s decision on the basis that the doctrine of res inter alios acta did not apply because the loss had been avoided by the repayment of the claimant’s loans as a result of Mr Hunt’s refinancing of EMSL. The loss arising from the claimant’s loan to EMSL had been made good when it had been repaid to the claimant by the borrower, and the fact that the money used to repay the loan had been borrowed from Mr Hunt was no more relevant than if it had been obtained from a bank or some other unconnected party. The payment made by Mr Hunt to the borrower and then by the borrower to the claimant to pay off the loan could not be regarded as collateral since the transaction discharged the very liability the existence of which represented the claimant’s loss.
168. Mr Sinai submitted that the Supreme Court did not, however, dispute the Court of Appeal’s description of the legal principles applicable to collateral benefits. He relied on the judgment of Lord Sumption (with whom Lords Neuberger, Clarke and Hodge agreed), where he stated at [11]:
“The general rule is that loss which has been avoided is not recoverable as damages, although expense reasonably incurred in avoiding it may be recoverable as costs of mitigation. To this there is an exception for collateral payments (res inter alios acta), which the law treats as not making good the claimant’s loss. It is difficult to identify a single principle underlying every case. In spite of what the latin tag might lead one to expect, the critical factor is not the source of the benefit in a third party but its character. Broadly speaking, collateral benefits are those whose receipt arose independently of the circumstances giving rise to the loss. Thus a gift received by the claimant, even if occasioned by his loss, is regarded as independent of the loss because its gratuitous character means that there is no causal relationship between them…”
169. Applying these principles to the present case:
(1) The cost of the API (other than the 68.1 grams used in clinical trials) could not be avoided since it is loss which had to be incurred to manufacture the new validation batches, as evidenced by Recipharm’s Validation Protocol;
(2) Payment by CompLex is clearly not mitigation by the Claimant since it was not a transaction that the Claimant could have brought about by itself;
(3) It is wrong to suggest, as the Defendant does, that the Claimant has not suffered loss. As Sales LJ stated the issue is basic justice between the person who has suffered the loss (the Claimant), the person who is in law responsible for causing the loss (the Defendant) and the third party who has made a payment which reduces that loss (CompLex). The law approaches the question on the basis of justice, reasonableness and public policy;
(4) In Swynson, the claimant was seeking damages for an unpaid loan made on the strength of the defendant’s breach of duty. Mr Hunt’s refinancing and repayment of the loan resulted in the very loss being claimed being repaid and extinguished. Unlike the transaction in Swynson, payment by CompLex for the API did not, by its intrinsic nature, extinguish the Claimant’s loss in having to redo process validation through Recipharm. Instead, payment by CompLex has benefited the Claimant collaterally in an amount equivalent to the loss which the Claimant has incurred, but not satisfied that loss.
(5) As Lord Reid held in Parry v Cleaver, the distinction between receipts which must be brought into account and those which must not must depend not on their source but on their intrinsic nature. Or as Lord Sumption put it in Swynson, the critical factor is not the source of the benefit in a third party but its character. Mr Beckers explained in his evidence that buying the API itself would have brought the Claimant into big financial difficulties as the funding was not there and that is why CompLex decided to help out ;
(6) The payment by CompLex cannot be said to have been made in the ordinary course of business. The Claimant had already paid circa US$1m in August 2012 for the API which the Defendant used to manufacture 12 validation batches. The subsequent payments by CompLex were unforeseen purchases of API to remanufacture process validation, which was not otherwise required, and which was lost. The Defendant did not suggest that CompLex made the API payments in the ordinary course of business.
170. In short, other than to put the Claimant to proof in paragraph 33(ii) of the Defence, the Defendant has not identified any good reason based on justice, reasonableness and fairness which should discharge it from accounting for the consequences of its breach of contract by reason only of CompLex having helped out.
171. The fact that CompLex paid the invoices or that the Claimant and CompLex were not in the same group is beside the point; the payment of the invoices by CompLex is the collateral benefit to the Claimant. The Defendant’s error is in focusing on the source of the payment. However, as stated by the Court of Appeal and by Lord Sumption in the Swynson case, the critical factor is not the source of the benefit but its character (or its “intrinsic nature” ).
172. I find that the Claimant is entitled to be compensated for the loss that is directly attributed to the Defendant’s failure / inability to complete its obligations under the DA, the ambit of such obligations I have set out above. At the time the DA was entered into it was envisaged that the Defendant would be the manufacturer and the steps that had to be taken by the Defendant included performing the Development of Product and co‑operating with the Claimant to achieve marketing authorisation for the Product. The fact that both parties were taking a commercial risk in not having a Supply Agreement in place, does not prevent recovery of the costs incurred by the Claimant in being able to ensure the fulfilment of the Defendant’s obligations under the DA by a third party, giving it the benefit of the bargain entered into. There was no guarantee that market authorisation would necessarily be granted, or, that if it were, that the Defendant could have insisted on being the manufacturer of the Product going forward, or that the Claimant could have insisted that the Defendant agree to fulfilling that role. As stated at paragraph 149(2) above, it was common ground that if a new manufacturer were to be chosen, that the process validation would have to be redone. That does not, however, in my judgment preclude the Claimant recovering the costs which it has incurred in completing the Defendant’s obligations under the DA, which it was unable to fulfil, and the additional expenditure related thereto, in order to bring the MAA to a determination.
173. For the avoidance of doubt, I do not accept the Defendant’s submission that the evidence of Dr Strieder referred to at footnote 30 above, supports the submission that the Defendant was not under an obligation under the DA to produce a Module 3 draft. As earlier indicated at paragraph 68(4)(i) above, there was one additional work order that related to one small aspect of the Module 3 draft, which was the subject of an additional payment, but this did not mean that the remainder of the work required for the Module 3 draft was not included within the Defendant’s obligations under the DA.
174. In my judgment, despite Ms Tan’s evidence given on Day 3, p24, lines 10-15, the losses incurred by the Claimant included the production of the emergency batches. I refer to paragraph 36 above and the Defendant’s own risk assessment carried out in about mid-2014, referred to at paragraph 43 above and which included the passage:
“ Patients who have been recruited into clinical study, and who respond positively, are maintained through compassionate supply. This continued compassionate supply is diluting the availability of previously manufactured stock and this reducing the supplies available to continue active recruitment….”
175. I agree with the Defendant that in the absence of any obligation upon the Claimant to reimburse CompLex, a company which was at the material time not part of its group, or the employers of Ms Tan and Ms Schuller for the work claimed as additional expenses under paragraph 55(E), the Claimant has suffered no loss and therefore is not entitled to recover anything in this regard. The key difference between this case and Swynson is that the claimant in Swynson had incurred the loss (in respect of a loan), but this loss had then been extinguished (by a further subsequent loan). Here there is no loss to the Claimant: it has not incurred the costs or paid the sums sought at paragraph 55(A)(ii) and (E) the amended Particulars of Claim and is under no obligation to pay CompLex.
176. I do not accept the Claimant’s analysis that this is the wrong focus and that CompLex’s payments are to be regarded as collateral benefits, such as not to prevent recovery of these sums by the Claimant. Had CompLex made a gift to the Claimant directly, rather than paying Chirogate’s invoices, there is a good argument that such a gift would not prevent the Claimant maintaining its claim for the sums incurred in respect of the Chirogate invoices had it paid them. This would fall into the category described by Lord Sumption, where he said: “
“Thus a gift received by the claimant, even if occasioned by his loss, is regarded as independent of the loss because its gratuitous character means that there is no causal relationship between them…”
That did not happen here, however. CompLex itself paid the invoices, extinguishing any loss sustained by the Claimant and in the absence of there being any obligation contractually to repay that expenditure, I do not regard it as recoverable. The fact that matters may have been capable of being arranged differently, which may have entitled recovery is nothing to the point. As Lord Neuberger said in the Swynson case at [100]:
“The fact that a transaction could have been differently arranged does not mean that it must have the same consequences as if it had been differently arranged. As a matter of logic, such a proposition would lead to an impossible situation, and as a matter of experience, it is by no means unusual to encounter cases where a transaction could be structured in two (or more) different ways, each of which would have different consequences - both in law and in commercial reality.”
177. The claim in relation to costs incurred in relation to the work done by Ms Tan and Ms Schuller are irrecoverable on the same basis. They were not sums that the Claimant was and is under any obligation to pay and it has therefore suffered no losses in that regard.
178. For the reasons given above, I find that the Defendant was in breach of the DA and the Claimant’s claim succeeds on liability.
179. Having set out the principles on which the damages fall to be calculated, I invite the parties to consider whether an agreement can be reached on a figure for quantum and interest. In the event that it cannot, the parties should serve written submissions by 9am on Monday 24 April 2023, setting out their respective calculations and the reasons therefore, in particular it would be helpful if the Claimant could address any outstanding points relied upon by the Defendant set out at paragraph 157 above. I will then list this for a further hearing of one day on Thursday 27 April 2023, being a date convenient to both parties and their legal representatives. At that time, I will also deal with any consequential matters arising from this judgment, and these should also be addressed in the skeleton arguments.
180. It only remains for me once again to thank Counsel for their helpful submissions. I would also offer my sincere apologies for the delay in handing down this judgment, which in significant part has been caused by personal issues, resulting from serious family illness.
