1. This patent action began with a claim by the Claimant (“MSD”) to revoke European Patent (UK) 2,676,679 (“the Patent”). The Defendant (“Wyeth”) counterclaimed for infringement on a quia timet basis. MSD denied infringement and accordingly the trial proceeded with Wyeth opening as the patentee.
2. There was also an unconditional application to amend the Patent. Where I refer to claim numbers below I mean the claims as numbered in the proposed amended form.
3. The issues narrowed as the case went on.
4. At trial, the infringement issues were:
a) A point of construction on claim 1: is it limited to vaccines having polysaccharides of precisely the 13 specified serotypes, or does it cover vaccines with those 13 and more? Since the alleged infringing product (“V114”) has polysaccharides for 2 additional serotypes on top of the 13, there would be no infringement if the claim were so limited. This also applied to claim 16 and its dependent claims.
b) On claims 4/19, Wyeth alleged infringement by equivalents: those claims require polysorbate 80, whereas V114 contains polysorbate 20.
5. MSD alleged invalidity on the following grounds:
a) Anticipation of claims 1 and 16 by WO 2006/110381 (“Hausdorff 381”), which is a novelty-only citation. While not formally accepting its invalidity, Wyeth did not defend claim claim 1 (a product claim) against this attack, but the claim still had to be considered because other claims are dependent on it. On claim 16 (a use claim), the sub-issues were the applicable legal standard, and whether the required use to inhibit silicone induced aggregation was clearly and unambiguously disclosed, it not being disputed that the physical features of the required formulation were disclosed (being the same as for claim 1). This attack was not run against claims 2 and 17 or claims dependent on them, because those claims require a surfactant and Hausdorff 381 has none.
b) Obviousness over “Present and Future of the Vaccination against Pneumonia”, Pediatrika 2004; 24(4): 147-155 (“de la Pena”).
c) Obviousness over WO 03/009869 (“Chiron”).
d) Insufficiency, as explained further below, primarily as a squeeze against obviousness.
6. Also, MSD opposed the proposed amendment to the Patent in respect of claims 2 and 17 on the ground of added matter, on the basis that the limitation to a surfactant generally would be an impermissible intermediate generalisation.
7. Both obviousness attacks depended on very heavily disputed aspects of the common general knowledge (“CGK”).
8. There was a rather complicated relationship between some of the more subsidiary issues, in the following way:
a) The non-infringement argument based on the 13 serotypes point applies to all claims and so if MSD were to win on that there is no infringement at all.
b) Similarly, the main thrust of the obviousness case on either citation (using a surfactant to prevent silicone-induced aggregation in a formulation of the 13 serotypes) would knock out all the claims.
c) However, if MSD lost on the 13 serotypes infringement point and the obviousness case failed, it would have an alternative route to winning by (i) knocking out claim 16 for lack of novelty over Hausdorff 381; and (ii) fighting off the amendment to claims 2/17 for added matter; and (iii) defeating Wyeth’s allegation of infringement of claims 4/19 by equivalence.
9. An unusual feature of the case is that Wyeth made clear that it would not seek an injunction if it won, but only financial compensation. I do not know the motivation for this but in any event it is irrelevant to anything I have to decide.
10. There are two aspects of the procedural history of the proceedings that need to be mentioned.
11. First, at the CMC disclosure was ordered in relation to certain specifically defined issues. These included issues concerning an allegation of classical insufficiency, which in the event was not pursued at trial, relating to a presentation made by two of the inventors in Berlin in 2007. Disclosure was also sought relating to specific events during the making of the alleged invention, and in the course of successfully resisting this Wyeth’s Counsel submitted that evidence about the making of inventions was generally not useful in patent cases. But no assurance was offered about whether Wyeth would lead such evidence or not, and Wyeth was not put to an election such that it would have to give disclosure about the making of the alleged invention if it wanted to call evidence in support of its case on obviousness.
12. This became significant because in due course, and as I explain below, Wyeth served a witness statement from a Dr Khandke, one of the inventors, which covered the Berlin insufficiency but also went much wider and related to many aspects of the making of the alleged invention. Wyeth did not volunteer any additional disclosure and MSD did not seek any.
13. Wyeth’s skeleton argument for trial then relied on the evidence of Dr Khandke for non-obviousness generally, and in a short oral opening on the first day of trial Mr Hinchliffe QC, who appeared for MSD leading Ms Moggridge, objected that sufficiently full disclosure had not been given and to what he called the repurposing of the fact evidence to support non-obviousness generally. But he did not apply to exclude the evidence or for more disclosure. At the same time, the Berlin insufficiency was dropped.
14. In MSD’s written closing, it was then quite rightly accepted that it had prepared for trial on the basis that it would need to challenge at least some aspects of Dr Khandke’s evidence relating to obviousness. It was also quite apparent that it had planned to deploy some of her evidence in its favour on the issue. So its real complaints were that it had had to prepare extra cross-examination shortly before trial, and that disclosure was incomplete. In my view at the time of closings, both of these were true, but only to a very limited extent. It seemed to me then that Dr Khandke’s evidence had been very thoroughly tested, and although a couple of examples of allegedly incomplete disclosure were given they were inconsequential.
15. On the other hand, Wyeth should have been more forthcoming about its intention to rely so extensively on Dr Khandke’s evidence on non-obviousness, and to engage proactively with whether further disclosure was necessary and appropriate.
16. I return to this in relation to Dr Khandke’s evidence, and in doing so also address a more significant shortcoming in Wyeth’s evidence and disclosure that came to light after trial.
17. The second procedural matter concerns the question of proving the functional feature of claims 1 and 16, namely inhibition of silicone induced aggregation.
18. Wyeth was concerned that the PPD did not adequately cover this for the purpose of addressing infringement, while MSD had concerns about the implications for validity, and a hearing took place on 27 March 2020. Birss J made an order which contained the following recital:
“Upon the Claimant and Defendant by their Counsel accepting and agreeing, in respect of each of the claims of the patent in suit, that if the physical features of that claim are satisfied, then the functional technical feature (i.e. inhibition of silicone induced aggregation of a polysaccharide-protein conjugate) is satisfied for that claim.”
19. This recital had the effect that the functional feature of claim 1 and of claim 16 was taken in fact to be met by V114, and also that it was met for Hausdorff 381 (which by the time of trial was effectively accepted by Wyeth to have all the physical features, though it made no formal concession). That meant that no disclosure was needed on those issues, from either side.
20. There was however a lack of clarity at the start of the trial about the recital’s implication for the validity of claim 16 as a use claim. Mr Tappin QC for Wyeth (for whom Mr Lykiardopoulos QC also appeared) argued that a use claim could not be anticipated merely because the implementation of the prior art would inherently achieve a functional purpose. He said that there had to be disclosure of the objective and its achievement, and that the undertaking given to Birss J was not about the disclosure of the prior art.
21. I understood Mr Hinchliffe to accept this in the end, and in any event I agree with it. Birss J was concerned with the fact of whether the effect was achieved, not the disclosure of the prior art. It was only the former that arose in relation to the application that was before him.
22. Each side called two expert witnesses: a vaccinologist and a formulator.
23. In addition, Wyeth called Dr Lakshmi Khandke, whom I have already mentioned, as a fact witness. She spoke to formulation aspects of the making of the invention including how Wyeth identified the problem of silicone induced aggregation and how it then addressed the issue.
24. Wyeth called Prof Juhani Eskola, who retired in 2018. Prior to that he had a long and distinguished career in industry and in academia which included important work on pneumococcal vaccines and led to significant publications, which came up in the evidence.
25. He was an excellent witness, clear and focused in his answers to questions.
26. MSD called Prof Peter Seeberger, who has since 2009 been Director at the Max Planck Institute of Colloids and Interfaces in Potsdam, and Professor of Chemistry at the Free University of Berlin.
27. He had vaccine experience prior to 2006 in relation to malaria and Leishmaniasis, but only since 2010 has he had any specific experience with pneumococcal vaccines, so he had to rely on reading literature in order to put himself in the position of the ordinary skilled person at the priority date in relation to that.
28. On that basis, Wyeth submitted that he was sub-optimally qualified, and less well placed than Prof Eskola, to give evidence about the choice of serotypes for inclusion in a pneumococcal vaccine at the priority date. My assessment is that by drawing on his more general pre-priority experience, his experience since 2010, and his reading for this case he succeeded in putting himself satisfactorily in the position of the ordinary skilled vaccinologist at the priority date. So I reject Wyeth’s submission.
29. Prof Seeberger was also clear and direct in his answers. Overall he was an excellent witness.
30. It is therefore true that Prof Eskola had more relevant and more direct experience in the field and at the priority date than did Prof Seeberger, but as is well established the expert’s precise approximation to the notional skilled person is not the key question. Each was able to help me to identify the common general knowledge and the attitude and approach of the ordinary skilled person in their field.
31. In any event, the disputes between them were very limited and insofar as they differed in their opinions they each were saying what they genuinely thought.
32. Unlike the vaccinologists, I had real reservations about both formulator expert witnesses. I will explain the nature of my reservations and the reasons for them in some detail, because my assessment of the formulator expert evidence and my general preference for MSD’s expert over Wyeth’s is central to my decision on obviousness, which in turn is one of the central issues leading to my overall conclusion against Wyeth.
33. Wyeth called Dr Geert Vanden Bossche. Dr Vanden Bossche started as a vet, later moving to SKB in 1995 where he had a role in quality control, including stability, extending to vaccine products. He took on roles of increasing seniority but still relating to project management for vaccine work, covering both formulation and adjuvant aspects. This trajectory continued during and after the merger with Glaxo to form GSK.
34. He left GSK in 2006 to go to Novartis, which had recently acquired Chiron, a leading vaccine company, and he was based in California and Siena, Italy. He was Head of Adjuvants and Project Manager on a respiratory syncytial virus vaccine project.
35. In 2007 he moved to Solvay, as Global Project Director of Influenza Vaccines with overall responsibility for the adjuvantation and preclinical development of Solvay’s cell-based influenza vaccines.
36. In 2008 he went to work at the Bill and Melinda Gates Foundation, and he has had a number of roles in industry and non-profit organisations since then, but which I will not list since they are of dwindling relevance to assessing him as a witness.
37. In parallel with his work in industry, Dr Vanden Bossche has held academic positions relating to virology and immunology. They do not appear to have had real bearing on the issues in this case.
38. I had four areas of concern about Dr Vanden Bossche’s evidence.
39. The first was his manner of giving evidence. He was argumentative almost throughout. He gave many very long answers which did not address the question or progress the discussion. He was unable to see or consider the opposing viewpoint, and he was unwilling to qualify his evidence even where it was apparent that that was needed.
40. The second was his experience. Although his job titles in the roles I have mentioned above extended to formulation they covered many other things as well, including in particular project management and adjuvantation. I came to doubt his real experience in the formulation aspects relevant to this case. One example was that he could not recall relevant aspects of the formulation of two vaccines in development at Chiron (Novartis) while he was there. Although his tenure there was fairly brief, it seemed very odd that he did not know these things (and had not refreshed his memory) if he really was close to that work.
41. Looking at his CV, to which he was taken in cross-examination, the speaking invitations which he listed, for example, relate either to general topics of vaccine development or, where it is possible to identify a particular aspect of technology, to adjuvants, which I think is where his real experience mainly lies. His exposure to the formulation issues centrally relevant to this case at trial was, I conclude, peripheral to his jobs and superficial. This may be explained by the fact that prior to amendment claim 1 of the Patent focused much more on the aluminium adjuvant and did not contain reference to the use of a surfactant.
42. Another example which may illustrate his relative lack of exposure to the formulation issues relevant to this case, and which relates to the third area of concern, was his insistence that when using an aluminium adjuvant the skilled formulator would be absolutely confident that no aggregation was possible because of the stabilising effect of the adjuvant. He maintained this view despite accepting that up to about 40% of the conjugated protein could remain in solution, and to the extent that he said the skilled formulator would consider it unnecessary even to test for instability that might arise. I found this extreme proposition, which was unsupported by any document, thoroughly implausible.
43. The third area of concern was his intransigence in defence of positions that he had taken. This was most notable in relation to the adjuvant stabilisation issue that I have just mentioned, but applied generally.
44. The fourth area of concern was his omission to support his opinions with citations. Other than his CV he exhibited no materials to his reports at all. As well as the adjuvant issue identified above, he also gave evidence orally about the ways in which he said aggregation might arise other than the hydrophobic interactions that were common ground. His evidence on this involved theories about hydrophilic attractions and other forces, which were not supported by documents (other than a passing reference to Brownian motion in Akers, which was not mentioned until closing) and were not even put to Prof Crommelin, who explained why he thought they were wrong in evidence in chief. These were speculative ideas that Dr Vanden Bossche should not have advanced without some proper support.
45. MSD made numerous other criticisms of his evidence which supported my impressions expressed above, but I do not consider it necessary to set them out further.
46. Overall, I would have severe reservations about relying on his evidence.
47. MSD called Prof Daniel Crommelin, from the Department of Pharmaceutical Sciences of Utrecht University. At the priority date he was Dean of Faculty in that department.
48. In 1995 he was co-founder of OctoPlus, a contract research company developing drug products including therapeutic proteins.
49. Wyeth did not significantly criticise the manner in which Prof Crommelin gave his evidence. Mr Tappin concentrated instead on areas where his direct experience was lacking, where it was alleged his experience was less than that of Dr Vanden Bossche, and on whether he was too heavily freighted with the attitude of an expert on therapeutic drug proteins rather than vaccines. I suspect that the reason for not criticising the manner of his giving evidence was a tactical one, since doing so could only have led to a comparison with Dr Vanden Bossche which would have been unfavourable to Wyeth’s position. Since criticisms were not made I will not dwell on it, but I have to say that I found Prof Crommelin argumentative and stubborn, though far less than Dr Vanden Bossche.
50. As to lack of experience, there is no doubt that there were some areas where Prof Crommelin lacked direct experience. The problem of assessing what those areas were was made unnecessarily much harder by the attitude and approach taken by Prof Crommelin and MSD’s advisers to duties of confidentiality owed by Prof Crommelin to his customers at OctoPlus.
51. Thus, Prof Crommelin refused on grounds of confidentiality to confirm whether or not he had done various things at OctoPlus. The questions were put in general terms such as whether he had worked on polysaccharide-protein conjugates there, or whether he had done agitation studies on aluminium-adjuvanted vaccines, and I found it very hard to believe that a simple yes-or-no about his experience (especially a no) would have compromised confidentiality; he was not asked for even the name of any client, let alone what they had engaged OctoPlus for.
52. Prof Crommelin remained resolute about not answering these questions, and although his demeanour was calm throughout it cannot have been comfortable for him. I was surprised that this problem had not been foreseen by MSD’s advisers (and said so), and more surprised later to be told that it had been, but that the advisers had not identified any possible solution, or canvassed it with Wyeth’s advisers. I expect that more foresight and explanation to the Professor about the issues and possibilities, such as sitting in private, might well have avoided the whole problem.
53. In any event, all that I can do is to assume that Prof Crommelin did not have any experience in the specific areas concerned. They were, however, fairly narrow, and were:
a) Whether he had worked on polysaccharide-protein conjugates. All he could say is that at OctoPlus he had “worked on some vaccines”.
b) Whether he had done agitation studies on aluminium-adjuvanted vaccines.
c) Whether he had ever added surfactant to an aluminium-adjuvanted vaccine formulation.
54. What I mean by “fairly narrow” is that they did not detract from his having worked on the general areas of vaccines, aluminium adjuvants (although to a relatively minor extent and only on animals), agitation studies and surfactants. He had not done work in some respects where those were combined in specific ways.
55. More generally, Prof Crommelin came primarily from a background of therapeutic proteins rather than vaccines (and this is probably why he had, apparently, not worked on polysaccharide-protein conjugates). I deal with the potential significance of this in connection with the skilled team, where I have rejected Wyeth’s submission that the two fields were strictly separated from one another. In any event I find that Prof Crommelin was able to explain the basic science of instability and especially aggregation, the effect of silicone and the use of surfactants, and other formulation matters less central to the case, more than adequately. I was satisfied that he was able fairly and properly to place those matters in the context of vaccines based on his reading and his own, albeit modest, experience of them.
56. Dr Lakshmi Khandke was called as a witness of fact. She worked for Wyeth (and its predecessor American Cyanamid) from 1991 to 2006. In 2006 she was Associate Director of Formulation Development, Vaccine Division. She left in 2017 and now works for PATH, a non-profit organisation, in its Center for Vaccine Innovation and Access. She has been involved in other parallel proceedings between the parties.
57. MSD submitted that Dr Khandke’s recollection was imperfect, that this was compounded by the documents she had relied on to refresh her memory having been picked by Wyeth’s lawyers, and that she gave long, non-responsive answers at times.
58. It is of course not surprising that Dr Khandke’s recollection has faded, but I thought overall it was very good. Although the documents for her consideration may have been picked by lawyers she clearly took responsibility for her own evidence. The leading example relied on by MSD on this point at trial was that in her first statement she positively said that she had not seen the FDA document by Amy Rosenberg (as to which, see further below), but then had to accept that she had. This was a very minor issue in my view, no more than a matter of emphasis on her part, although the document in question is important, for reasons I also set out below.
59. In just a few places I found her interpretation of documents unlikely. In particular, a document of 7 January 2005 (Khandke XX/18) gave the clear impression that Wyeth was aware of problems with silicone from a number of events prior to that, but she said it had not been. I think she was wrong about that, but at the time there was no malice or lack of honesty on her part.
60. In relation to the manner of her giving evidence, I agree that she gave long answers on some occasions but it was unimportant because in the great majority of instances she was short and to the point. She was perhaps just a little argumentative and prone to see matters from Wyeth’s point of view, but that is to be expected from a long-time employee who has spent quite some time on this dispute, here and abroad. In my view she was completely honest.
61. Dr Khandke’s cross-examination was extensive and thorough. There is one matter that was not put to her and should have been: a document was put to Dr Vanden Bossche in cross-examination bearing the date 2 October 2003 and the name of Dr Robert Seid, who was a senior colleague of Dr Khandke. The provenance of the document and its context was not at all clear. Although Dr Khandke was not named on it she might well have been able to explain, or qualify it. On its face it seemed rather inconsistent with her evidence because it seemed to show an awareness of the problems of silicone well before they were experienced by Wyeth in the context of making the invention, and so it should have been put to her. That not having happened, I do not think I can rely on it.
62. Some weeks after the trial and when this judgment was in an advanced stage of preparation, Wyeth’s advisers found some documents which had previously been overlooked. They were directly relevant to Dr Khandke’s evidence and she made a fourth witness statement to explain what had happened. She restated and refined her position about the January 2005 document referred to above. In particular, she had to accept in the light of the new documents a vaccine referred to in the January 2005 document (whose name I will not include in this judgment because Wyeth says it is confidential, and that assertion will have to be assessed at the form of order hearing), had experienced aggregation problems which were attributable to silicone, before the project which led to the Patent (though she maintained that this was not the problem referred to in the document). In this respect, which was material to the issues, her evidence at trial was avoidably and regrettably wrong.
63. I received detailed written submissions about the new documents, from both sides. My conclusions are that:
a) It is unsatisfactory that the documents were not provided by Wyeth in time for trial. The details can be explored in relation to costs, as appropriate.
b) The documents undermine the reliability of Dr Khandke’s evidence in some significant respects but I remain of the view that she has not been malicious or dishonest. It is much more likely that the failing is that of Wyeth and its advisers.
c) The documents might well have been of use to MSD at trial to improve its case on silicone causing aggregation as being CGK, but it won on that (and on the Wyeth story) anyway, and I already agreed with it about the January 2005 document so I think there has been little or no harm done.
d) The episode exacerbates my concern about the way that disclosure about the Wyeth story came into the case and was managed. The fact that it avoided a rigorous regime for the giving of such disclosure is no excuse for missing the documents which have now been provided, or for allowing its witness to give evidence that was wrong.
e) But at least it drew them to MSD’s and the Court’s attention when found.
64. MSD served a CEA notice to introduce a statement of a Mr Baney of the syringe company Becton Dickinson. Nothing turned on this.
65. There was a basic dispute about the skilled team: would it include a vaccinologist? MSD contended that it would not and Wyeth argued that it would.
66. This was really a forensic point by MSD arising from its contention that the Patent contained no technical contribution relating to the choice of serotypes. It said that it followed that the Patent would therefore not be of interest to a vaccinologist.
67. I have rejected the lack of technical contribution argument, for reasons given below, but in any event the Patent would be of interest to a vaccinologist and a vaccinologist would be needed to implement it. It is true that claim 1 as proposed to be amended requires a specified 13-valent (hereafter “13v”) pneumococcal vaccine and an aluminium salt adjuvant, so the high-level decisions in these respects would be limited. But the vaccinologist would still have to attend to the details, and the potential inclusion of other types of polysaccharides (e.g. meningococcal) would have to be considered.
68. It was also not in dispute, as I understood it, that in real teams in this field there was a vaccinologist in the team. In addition, the prior art and de la Pena in particular is addressed to a vaccinologist, at least in part.
69. Wyeth made the somewhat cheap point that it was inconsistent with MSD’s position even to call a vaccinologist, but of course MSD had to do this in case its argument on lack of technical contribution failed and it had to rely on the obviousness side of the squeeze.
70. I therefore conclude, without hesitation, that a vaccinologist would be included in the skilled team. It was common ground that a formulator would be included.
71. I turn to the more detailed characteristics of the team and of each team member.
72. As to the team, there were relatively few real-world teams in this field and the design of vaccines was reserved to a small number of very specialist companies. In addition, they did not publish very much.
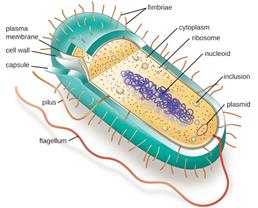
73. As to the formulator, there was a disagreement over the extent to which their knowledge and experience would relate exclusively to vaccines, or extend to more general matters of formulation and in particular to therapeutic proteins. This dispute also affected the parties’ contentions about the common general knowledge, and I touch on it there, too. In short, I reject Wyeth’s contention that the skilled formulator would have experience or knowledge exclusively in relation to vaccines.
74. Vaccines have their special complications and difficulties, but vaccine antigens were often proteins (or proteins conjugated to other moieties) and the skilled formulator would therefore need to know about issues concerning proteins generally.
75. Looking at this in more detail, Wyeth raised three points to argue that therapeutic proteins were “a completely different ball game” from vaccines at a technical level:
a) The first was that in vaccines, immunogenicity is desired whereas in therapeutic proteins it is problematic and even dangerous;
b) The second was that in vaccines, protein antigens do not usually need to have the “right” or native 3D conformation to achieve their goal, whereas in protein drugs conformation is critical to efficacy, as it determines binding to the receptor;
c) The third was that in protein drugs the protein is the active ingredient but in polysaccharide-protein conjugate vaccines it is a carrier.
76. Each of these is true, but beside the point. Both in vaccines and in therapeutic proteins it would be equally unacceptable to have aggregation/precipitation and the skilled team would need to know how to deal with it if it happened. It would not arise, or be thought to arise, from the fine details of 3D conformation, being a much grosser problem, or to be related to immunogenicity. It would be understood to arise from the fact, common to protein drugs and vaccines with protein antigens, that one was dealing with proteins.
77. In saying this, I am not holding that the skilled formulator in the team would be able to function if they knew only about therapeutic proteins. They would need to know a lot more.
78. If vaccine formulation were such a specialised and isolated field as Wyeth alleges one would expect to see much more evidence of qualifications and career paths that involved exclusively and specifically vaccines. While some might have moved to vaccines very early in their careers, others such as Prof Crommelin would have worked on vaccines and other drug products.
79. As to the vaccinologist, there was broad agreement that the field is an applied art and those who work in it come from a broad range of backgrounds. It is not taught in universities as a specific subject. There was a dispute between the vaccinologist experts about the degree to which the notional vaccinologist would be interested in glycobiology, but this was a reflection of their individual interests and anyway did not turn out to matter.
80. There was broad agreement about the principles applicable. Wyeth referred me in its written opening to the judgment of Kitchin LJ (as he then was) in Idenix v Gilead [2016] EWCA Civ 1089 at [70]-[72] and the judgment of Arnold J (as he then was) in KCI Licensing Inc v Smith & Nephew PLC [2010] EWHC 1487 (Pat) at [104]-[112].
81. It was also common ground that (as was also pointed out in KCI, ibid.) there is a distinction between matters that are truly CGK and those that would be found by routine means as the skilled person set about a problem. CGK informs the skilled person’s work from the outset; information found by routine means may come later. Wyeth referred me in this respect to Generics v. Daiichi [2009] RPC 4. This principle was not disputed, but its application in this case was (if only by implication in the way that MSD ran its case), as will appear from my reasoning below in relation to the use of surfactants in known vaccines in particular.
82. Wyeth also reminded me of the statement of the Court of Appeal in Beloit v. Valmet [1997] RPC 489 and 494-495, referring back to dicta of Luxmoore J in British Acoustic Films (53 RPC 221 at 250) that “It is certainly difficult to appreciate how the use of something which has in fact never been used in a particular art can ever be held to be common general knowledge in the art”.
83. This may be an important principle where it applies, but in my view it depends on what the “something” that has not been done before is, and how it is characterised. In the present case, Wyeth contends that no one had previously used a surfactant to prevent silicone induced aggregation in an adjuvanted conjugate vaccine. But whether or not that was so, workers certainly had used a surfactant to prevent aggregation of proteins - that was accepted to be CGK - and they would not expect adjuvanted conjugate vaccines with their protein portions to be different in any relevant way to that. It is not real to say that something phrased very narrowly has not been done before, when it would be, from the skilled team’s perspective, merely a specific instance of doing something that was well known as expressed in broader terms.
84. It is also worth saying something about the sources that may be, and typically are, used to prove CGK. This is discussed in Terrell, 19th Edn. at 8-75 to 8-79.
85. CGK is often proved by means of textbooks. These have the advantage that their contents, scope and intended audience are readily ascertainable. Not all textbooks are well known and widely accepted, and that has to be proved by evidence, but frequently is not disputed.
86. The fact that textbooks are used in this way does not put a conceptual limit on what materials can be used, however. Individual publications in the scientific literature and patent specifications could represent CGK, for example, but it depends on evidence that they had sufficient reach, impact and acceptance. In general, they may be less likely than textbooks to represent CGK. Repeated individual publications of the same information (and as Floyd LJ has repeatedly said, it is information that one is focused on) may, with the right evidence, show that it was CGK, even if each of the publications on its own would not.
87. In the present case, MSD relies on documents such as syringe manufacturers’ commercial literature about reducing silicone, and an FDA document referring to silicone-induced aggregation as being a well-known problem. These individual documents were clearly not well known in themselves, and MSD does not say that they were. But I see no reason why they cannot in principle form part of the picture in assessing whether the issues around silicone were widely discussed, which in turn could support a conclusion that those issues were CGK.
88. The following technical background section on the CGK of the vaccinologist is an edited-down version of the relevant section of the primer.
89. The encapsulated bacteria Streptococcus pneumoniae (pneumococcus), Neisseria meningitidis (meningococcus) and Haemophilus influenzae type b (Hib) have been responsible for a significant proportion of severe infections in children for decades, specifically bacteraemia and meningitis. Encapsulated bacteria are therefore an important class of bacterial pathogens. These bacteria have potentially severe outcomes, such as sepsis, pneumonia, and meningitis.
90. Encapsulated bacteria are covered with an outer capsule or shell of polysaccharides. Polysaccharides are carbohydrates.
91. Figure 1 below shows a drawing of a bacterial cell with a polysaccharide capsule on the outside of the bacterium. Because the polysaccharide capsule is on the outside of the bacterium, the polysaccharide can be recognised and bound by antibodies of the (human) immune system.
92. Pneumococcal bacteria commonly colonise the human nasopharynx. Such colonisation does not usually result in any symptoms. However, if pneumococci are able to invade other sites in the body, for example the lungs, middle ear, meninges or bloodstream, they can cause severe disease that may result in death or disability. The most common diseases are otitis media (infection of the middle ear), bronchitis and more seriously, the organism can cause bacteraemia (invasion of bacteria in the bloodstream), meningitis and pneumonia. The most severe effects are on young children (less than five years of age) and those with weakened immune systems.
93. Within a species of encapsulated bacteria, "serotypes" or "serogroups" are used to classify the variations that occur in the particular polysaccharides displayed on their surface. For example, more than 90 serotypes of pneumococcus have been identified. There are geographical, temporal and demographic differences in the distribution of disease-causing serotypes. Further, the disease (and the severity of the disease) caused by each serotype may differ.
94. By the priority date of the Patent, the most prevalent and/or virulent serotypes of encapsulated bacteria, such as meningococci and streptococci (including pneumococci), affecting young children and other parts of the population were actively monitored by public health bodies in many countries (in the UK by the Health Protection Agency).
95. There are two types of immune response in humans - known as innate and adaptive immunity. Both feature humoral systems (i.e. mediated by macromolecules such as complement or antibodies) and cell-mediated systems (i.e. mediated by cells such as macrophages or killer T-cells).
96. The innate immune system includes all the systems which will react against an infection in a non-specific way.
97. The adaptive immune system may be activated when a pathogen eludes or overwhelms the innate immune system. The adaptive immune system develops in response to specific features of the pathogen - the antigens. The adaptive immune system is able to learn from previous encounters with pathogens and mount a much faster and more effective response when the same pathogen is encountered again. This phenomenon is referred to as immunological memory.
98. B-cells and T-cells are key components of the adaptive immune system. B-cells are lymphocytes that originate and mature in the bone marrow while T-cells are lymphocytes that originate in the bone marrow but migrate to the thymus to undergo maturation.
99. Antigens are substances that can be recognised by the body’s immune system. If a B-cell or T-cell receptor recognises an antigen there may be sufficient stimulation for the particular lymphocyte to initiate an immune response. An antigen that induces such an immune response is referred to as an immunogen. Although all immunogens are antigens, not all antigens are immunogenic. Antigens may have multiple immune recognition sites, referred to as epitopes.
100. T-cells are activated when an antigen presenting cell (e.g. a dendritic cell or a B-cell) displaying a peptide-MHC complex is recognised by a T-cell receptor.
101. B-cells can be activated by two different routes: helper T-cell-dependent and helper T-cell-independent activation.
102. When activated by an antigen by either route, B-cells produce antibodies which circulate in the blood. Antibodies are proteins which bind to antigens.
103. As antibodies circulate in the body, they recognise, bind and potentially neutralise antigens that are identical, or at least similar enough, to the one that triggered the immune response. By binding to the antigen, an antibody can tag an antigen, pathogen or an infected cell for attack by other parts of the immune system (e.g. killer cells or the complement system), or can neutralise its target directly.
104. A vaccine is an antigen-containing preparation that provides acquired immunity against a particular disease. It prevents infectious diseases by priming the immune system prior to exposure to disease-causing organisms (i.e. pathogens), such as bacteria or viruses. Vaccination induces a type of immunity that is provided by natural infection without causing the symptoms and complications of the disease.
105. Vaccines might include an attenuated/inactivated version of the pathogen (to avoid causing disease), a part of a pathogen (e.g. purified membrane components, purified proteins, purified polysaccharides) or a recombinant pathogen-derived protein as the antigen.
106. When stimulated by a vaccine the immune system activates immune effector cells (e.g., B-cells and T-cells) in response to the antigen(s) being introduced into the body. In this way, vaccines “teach” the immune system to specifically recognise certain antigens, giving it the ability to react quickly upon future pathogen exposure due to the immune memory generated by the vaccine. Vaccination thus, in theory, provides immunological protection prior to infection against pathogens from which vaccine antigens are derived.
107. When the source of an infection is a species of encapsulated bacteria, the immune system often directs its response to the polysaccharides of the capsule. This makes these polysaccharides attractive antigen molecules for vaccines.
108. By the priority date of the Patent, polysaccharides had been successfully used as vaccines in adults and older children against, for example, pathogens such as pneumococci, certain meningococci and Hib. In a polysaccharide vaccine, only the sugar part of the bacteria (i.e. the polysaccharide capsule) is included as the antigen to stimulate the immune response. Polysaccharides induce a helper T-cell-independent antibody response.
109. One such example is the Claimant's Pneumovax 23 vaccine - a 23v pneumococcal vaccine authorised in the UK in 2000 and still on the market today. It includes polysaccharides of pneumococcal-serotypes 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19F, 19A, 20, 22F, 23F and 33F.
110. Antibodies can be serotype-specific, recognising the specific structure of that polysaccharide. Antibodies against a polysaccharide from one serotype may not be cross-protective (or may only be weakly cross-protective) against different serotypes. Because of this lack of cross-protection, vaccines may be multivalent, i.e. they include polysaccharides from more than one serotype.
111. There may be progression in the development of multivalent vaccines in relation to the number of serotypes used. The earliest version would generally utilise the most prevalent and most virulent serotypes. Over time, later vaccine versions will incorporate additional clinically-relevant serotypes for broader protection when a sufficient clinical need is identified and considerations such as manufacturability, marketability and cost allow.
112. Although bacterial polysaccharides were successful as antigens in vaccines for immunisation of adults and older children against bacterial infections, this was not the case for young children, particularly those under 2 years of age, who produced a very poor immunogenic response. This is the section of the population which is most prone to infection due to the fact that their immune system is still developing.
113. As early as the 1920s, it had been shown that the antibody immune response to the polysaccharide capsule of bacteria could be improved by conjugating polysaccharides with carrier proteins. In such a conjugate, the polysaccharide antigens are bound to a carrier protein. Common carrier proteins for such polysaccharide-protein conjugates were tetanus and diphtheria toxoids, the outer membrane protein complex of Neisseria meningitidis serotype B, and CRM197 (an abbreviation of "Cross-Reacting Material 197" - a non-toxic mutant of diphtheria toxin with only a single amino acid change).
114. In a conjugate vaccine, the polysaccharide and the carrier protein each stimulate the immune response. Through conjugation to carrier proteins, a robust antibody-mediated response against the polysaccharides can be achieved.
115. Conjugate vaccines have proven to be much more effective than polysaccharide vaccines in young children (they are also efficacious in adults).
116. By the priority date of the Patent, conjugate vaccines which were effective against invasive bacterial diseases in infants had already been available for a number of years. The first commercially available conjugate vaccine - a Hib conjugate vaccine - was first marketed in the USA in 1987 and was introduced into the infant vaccine schedule in the UK in 1992. Formulations of Hib conjugate vaccines authorised before the priority date used various carrier proteins, including carrier protein CRM197.
117. The Defendant's Prevenar® (or Prevnar®) vaccine, a 7v pneumococcal conjugate vaccine, was the only authorised protein conjugate vaccine for pneumococcal disease at the priority date. It was approved for the US market in 2000 and the EU in 2001. Prevenar® contains polysaccharides of seven pneumococcal-serotypes: 4, 6B, 9V, 14, 18C, 19F and 23F, each conjugated to CRM197.
118. Adjuvants are substances that, when associated with an antigen, increase the antigen's ability to provoke an adaptive immune response - literally vaccine "helpers". It has been known since the 1920s that the immunogenicity of an antigen can be enhanced by using an adjuvant.
119. The most widely used adjuvants in human vaccines are aluminium salts and it was well known that they boosted immunogenicity by adsorbing protein-based antigens. At the priority date, aluminium salts were the most common adjuvants in approved human vaccines in the US and the UK. Prevenar® used aluminium phosphate as an adjuvant.
120. The preceding two paragraphs were included in the primer in a section about formulation, but they in fact relate to the immunological function of adjuvants. There are also formulation implications of adjuvants and I address them below. Both members of the skilled team would therefore be interested in whether an adjuvant was used, and which.
121. That concludes the matters that were contained in the primer as to the CGK of the vaccinologist. As may be seen, there was a lot of agreement. But there are other relevant matters on the topic where there either was a dispute, or comment is needed, and which I will now address.
122. At the priority date, it was, it is common ground, CGK that it would be desirable to add more serotypes to pneumococcal conjugate vaccines. The reasons would include, in particular, adding serotypes for which cross-protection from those already in Prevenar (for example) was either weak or absent.
123. It was also CGK however that there were practical limitations to doing that, and that a choice had to be made of which serotypes to try to include.
124. Further, it was CGK that various 9v pneumococcal vaccines were in development, which would add serotypes 1 and 5. But at the same time, it was CGK that two 11v vaccines, from Aventis and GSK, which would add serotypes 1, 5, 3 and 7F to Prevenar’s list, had failed, for different reasons (Aventis had given up and GSK had dropped down to 10v, omitting serotype 3). The precise reasons for failure do not matter, save to say that they were specific to the efforts of those companies, and not regarded as reasons why that combination of serotypes could never be added.
125. Against this backdrop, MSD submits that it was CGK that the overall direction of travel was to increase the number of serotypes. In my view this is much too simple and is symptomatic of MSD’s obviousness case over Chiron (de la Pena is quite different) being, on the vaccine side at least, really a case over common general knowledge alone, and suffering from the common vice in such cases of using a level of abstraction divorced from the problems of the real world. It is true that the skilled vaccinologist would not progress a vaccine that left out serotypes which were present in Prevenar (or whatever the state of the art starting point might be), but that does not mean that they would be wedded to adding further serotypes, let alone multiple serotypes, or that they would think that any combination with added serotypes could easily be achieved without problem. They would know that there could well be problems.
126. Similarly, 11v pneumococcal vaccines were not common general knowledge in the usual sense of being widely accepted as a good place to start. On the contrary, they had failed. MSD point out that, as I have said, they had failed for reasons specific to those workers, but that is not the same as there being a confidence that they could readily be done. So why, as a matter of CGK, would the skilled vaccinologist start from an 11v vaccine rather than the 9v? MSD’s case that they would do so injects hindsight into the CGK discussion, in my view.
127. A further aspect of the vaccinology CGK that I must address relates to the knowledge around certain specific serotypes, in particular 6A and 19F. These are the focus because it is by adding those two to the experimental 11v vaccines that MSD seeks to reach the 13v combination required by the claims of the Patent, at least in the obviousness case over Chiron.
128. It was essentially common ground that it was CGK that the existing serotypes did not give cross-protection for 19A and that, for reasons that I need not go into in any greater detail, 19A was an important serotype clinically. I can and do accept that in principle that is the sort of thing that could be CGK: not that 19A could necessarily be added with ease in practical terms, but that immunologically speaking it would, if it could be added, provide important benefits.
129. By contrast, there was a definite and lively dispute about the art’s attitude to serotype 6A, and whether cross-protection against it was achieved by the existing inclusion of a serotype 6B antigen.
130. Although the parties addressed this as a matter of CGK, I am not at all sure that it was. The cross-examination went into the fine detail of numerous papers; detail which I felt went well beyond that which could reasonably be expected to be CGK. It felt much more like an obviousness case based around documents which might, but not necessarily would, be found by routine means. It is often difficult to distinguish this from CGK, of course, but it may be necessary to do so and it helps to articulate what one is dealing with. If it were only an argument about what actually was truly CGK I would certainly hold that MSD failed to establish that it was CGK that serotype 6A was in fact necessary to make up for a lack of cross-protection from 6B.
131. Dealing with the issue as one of information that would be found by the skilled person taking obvious steps from a specific starting point, I do not find it necessary to go into the details of all the papers that were put on the 6A/6B issue. Profs Eskola and Seeberger had genuinely held but opposing views about the utility of 6A. My finding is that there was evidence in both directions and that the notional skilled person would consider, and it was CGK, that while a case for the utility of 6A could be made, the situation was still developing. It is not possible, or necessary to this conclusion, and the evidence was not detailed enough, to identify exactly which papers the skilled vaccinologist would find and work through.
132. The cross-examination of Prof Eskola proceeded consistently with that: it was put to him that it would not be “irrational” to include 6A and he agreed. That is simply not the same thing as saying that it was CGK that 6A ought to be included in the next round of additions, or would give essential cross-protection.
133. Similarly, it was put to Prof Eskola that 6A and 19A were “either at the top of, or close to the top of, the list of serotypes that were causing disease, but that were not in a vaccine yet?”. He said that the reason they were not in a vaccine to date was because the art thought there was adequate cross-protection. Even if he had not qualified his answer in that way, the mere fact that 6A and 19A were strong candidates (clearly the former less than the latter) does not elevate their necessary or imminent inclusion to CGK.
134. I should make it clear that in my view these criticisms of MSD’s case come home to roost in relation to the attack over Chiron. The position over de la Pena is quite different because, as I will explain, that included the information that a 13v pneumococcal vaccine including 6A and 19A had been put into trials by Wyeth with success.
135. Wyeth and Dr Vanden Bossche took a very negative approach to the CGK of the formulator, confining themselves mainly to attacking the CGK status of anything that might help MSD’s case. They gave much less effort to identifying what was CGK. This makes the task of providing an uncontroversial summary of areas of agreement very difficult. What I propose to do, in broad terms, is to set out that which was agreed, then to identify the points of dispute, and then to go through the main materials relied on by MSD as a whole explaining what they each show, in my view. This seems more practical and readable, since the materials tend to cover varying combinations of the topics.
136. The following matters were agreed, entirely or subject to the minor reservations mentioned, to be CGK.
137. Buffers are solutions which contain a weak acid and its salt, or a weak base and its salt, and can be used to control and maintain pH. This was common ground, as was the fact that it was desirable to have a buffer with the ability to keep a vaccine close to physiological pH (7.4). At the start of the trial it seemed like there might be an argument that histidine buffers were obscure or regarded as undesirable in some way, which might have affected the obviousness case over Chiron. But this faded away and in the end Wyeth accepted that it would be obvious to include a buffer if starting from the prior art.
138. It was also common ground that where an aluminium adjuvant was used (to boost the immunological effect of the vaccine) it would have the effect of stabilising of the protein present by adsorption to the adjuvant, preventing adsorption to any hydrophobic interfaces (as to which, see below).
139. I am not sure how far Wyeth contended that this would go; certainly it said that the skilled formulator would try to maximise adsorption to the aluminium adjuvant and I accept this (as I think did MSD) as CGK. But I understood Wyeth also to accept, and I find, that the CGK was that some proportion of the protein might well remain in solution, not adsorbed to the aluminium adjuvant. While the formulator would hope that this would not be enough to lead to any material adsorption or aggregation, it would be necessary to determine empirically whether that was so. Dr Vanden Bossche agreed that up to 40% of the protein might remain unbound by the adjuvant.
140. What certainly was disputed was Dr Vanden Bossche’s more extreme position that the skilled formulator would have such confidence in the mechanism of adsorption to the aluminium adjuvant that he or she would be satisfied that adsorption/aggregation was not possible, or so unlikely that it would not even be tested for. There was no support for this anywhere, it is not logical, and is inconsistent with his acceptance that there could be appreciable protein remaining in solution. I reject it. It is not the case and was not CGK.
141. A surfactant is a material which lowers the interfacial tension between two liquids, a liquid and a solid, or a liquid and a gas. Surfactants have a hydrophilic and a hydrophobic part. They can bind to hydrophobic interfaces.
142. MSD contends that the skilled formulator would know as a matter of CGK that surfactants could be used to prevent protein aggregation at interfaces, by binding to those interfaces in preference to the protein doing so. This is a central debate and I return to it below.
143. However, I understood it to be common ground that surfactants had other useful purposes in vaccine production, including at stages prior to the final formulation, such as solubilising cellular proteins and lysing cells in bulk preparations. This is significant because it means that a skilled person seeing the presence of a surfactant in a vaccine could not conclude that it was there to prevent aggregation or, more generally, as an excipient with a function in the final formulation. It might be a residue from earlier in the manufacturing process.
144. Proteins such as those used in vaccines (on their own or conjugated to polysaccharides) have hydrophobic and hydrophilic regions. In solution the protein folds so that its hydrophobic regions are inside and the hydrophilic regions are outside. This means that there is little or no opportunity for the hydrophobic regions of individual proteins to interact; if they could then they would tend to stick to each other.
145. When proteins come into contact with hydrophobic interfaces such as container walls (or air or oil interfaces) they may adsorb to them. This can cause a partial unfolding of the proteins, revealing some of their previously-internalised hydrophobic regions. These may then bind to each other, resulting in aggregation of multiple proteins. The aggregates can precipitate and form visible particles.
146. In this way, adsorption is the precursor to aggregation.
147. Dr Vanden Bossche suggested that some small degree of aggregation might be acceptable. Whether or not this was so was not really pursued with Prof Crommelin and in any event it was clear that a significant amount of aggregation was viewed as a bad thing, since it would lead to problems including batch-to-batch inconsistency and precipitation, and it was common ground that they were to be avoided.
148. Up to this point I believe what I have said was accepted to be CGK and in any event I find that it clearly was. But there was a dispute over an ensuing point of detail, as follows.
149. The mechanism of adsorption and aggregation explained above is based on hydrophobic interactions. Dr Vanden Bossche also referred in his written and oral evidence to the effect of electrostatic attraction, ligand exchange, van der Waals forces and hydrogen bonding as mechanisms for adsorption. He appeared to go quite a lot further in his oral evidence in these regards. These propositions were not really explored with Prof Crommelin in cross-examination, even though he had robustly rejected them in examination in chief after hearing what Dr Vanden Bossche had said. Their significance would have been to suggest that aggregation was not necessarily preceded by adsorption at an interface but might take place in the aqueous phase. At this level, Prof Crommelin was asked, and expressed the view, which I accept, that any aggregation not preceded by adsorption was very minor.
150. To put it another way, I think it was clearly established to be CGK that adsorption at an interface followed by aggregation, mediated by hydrophobic interactions, was at least the main mechanism that the skilled formulator would have in mind in relation to the issues in this case.
151. I have not lost sight of the fact that this case is about polysaccharides conjugated with carrier proteins, but it was not suggested that this would prevent aggregation, or make irrelevant the issues that I have just identified.
152. This was a central dispute and I will deal with it distinctly.
153. First of all, there was ample material in the case to justify the proposition that surfactants were known as a matter of CGK to be used to prevent aggregation of therapeutic proteins. Prof Crommelin gave numerous examples, and Dr Vanden Bossche accepted the proposition. Whether this meant that it was CGK to a vaccine formulator raises the issue about the identity and characteristics of the skilled person that I have dealt with above. Since I have held that MSD is right on this point, it follows that it was CGK to a vaccine formulator that surfactants could potentially be used where aggregation was encountered.
154. Second, Dr Vanden Bossche accepted that the skilled formulator would know as a matter of CGK about the use of surfactants to address aggregation in vaccines where an aluminium adjuvant was not used, for example because it could not be, as in a live virus vaccine. Of course, Dr Vanden Bossche took the view that that was a different situation because of the skilled formulator’s very high degree of confidence in the adjuvant stabilising all the protein present, but I have rejected that. In any case, the knowledge of using a surfactant in such circumstances establishes, in my view that it was CGK that a surfactant could be used in a vaccine where necessary, if aggregation was observed.
155. Third, the materials put forward by MSD show that the use of surfactants to address aggregation in vaccines generally, if it occurred, was CGK. I analyse those materials below.
156. The use of silicone in pre-filled syringes as a lubricant was accepted by Dr Vanden Bossche to be common general knowledge, and it clearly was. However, he did not accept that it was common general knowledge that it could cause problems, and in particular he did not accept that it could cause aggregation.
157. I find based on the materials identified below that it was CGK that silicone presented a hydrophobic interface.
158. The fact that silicone was known to be used in pre-filled syringes and that it could present a hydrophobic interface does not in itself necessarily mean that it was known, or would be expected, to cause problems generally, or aggregation specifically. But I hold on the basis of the materials that I analyse below, that it was.
159. There was a basic disagreement between the experts about whether the skilled formulator would have an interest in, or knowledge of, the container into which a pharmaceutical product would be filled. Prof Crommelin said that they would and Dr Vanden Bossche said they would not.
160. On this point I agree with Prof Crommelin. His view was supported by Akers (see below) and was also consistent with the evidence of Dr Khandke, and this was one of the areas where I found the specific experience in Wyeth to be of value as a cross-check on the CGK. It also makes sense: the skilled formulator’s basic CGK would be that, for example, the hydrophobicity of formulation components could be important. What justification could there be for ignoring that the container itself was hydrophobic? None, in my view.
161. I found the following parts of the textbooks and other materials to be of particular importance. That is not to say that they are only parts I considered, of course.
162. Akers is a 2002 textbook. Its title is “Development and Manufacture of Protein Pharmaceuticals” and it is about therapeutic proteins in general and not vaccines. I have addressed the non-significance of this above.
163. MSD referred in particular to Chapter 2, on which Akers, who was at Baxter, was the first author.
164. At page 52 Akers contains the following:
4. WHY PACKAGING, PROCESSING, AND FORMULATION
ARE INTERRELATED
Most publications that deal with protein formulation do not cover
aspects of the manufacturing process or packaging. Yet the three are
interrelated. A formulation is not stable unless the product can be
manufactured consistently at a large scale and packaged in a container/
closure system that can maintain sterility and stability for a relatively long
period of time. Packaging of proteins is especially challenging because of
the inherent interactive nature of proteins with inert surfaces such as glass,
rubber, and plastic. For many proteins, adsorption at these surfaces sometimes
results in the surface denaturation and subsequent aggregation
of the protein (Cleland et at., 1993).
165. And then at page 53:
The bottom line message here is simple: A formulation scientist
developing a protein (or, for that matter, any) dosage form must consider
the formulation, process, and package together, not focus on one aspect
exclusive of others. The smart formulation scientist, in fact, not only will
consider all aspects of the formula, process, and package, but also will develop
close interactions with packaging engineers, polymer scientists, manufacturing
experts, and other experts in areas outside of the formulation scientist's
direct expertise. ("None of us is as smart as all of us"-Satchel Paige.).
166. This supports Prof Crommelin’s and Dr Khandke’s view as mentioned above.
167. At page 69 it says this:
7. PHYSICAL STABILIZATION
Physical instability is rarely encountered in formulations of small drug
molecules except for poorly water-soluble compounds. Proteins, because of
their ability to adopt higher ordered structures, tend to undergo a number of
changes structurally, independent of chemical modifications. Physical
instability of proteins is often a greater cause for concern and may be more
difficult to control than chemical instability. Virtually all protein structures
have hydrophobic regions to some extent, and low solubility in water is
regarded as an indication that a material is hydrophobic (Currie and
Groves, 1992). Hydrophobic interaction is a major driving force for protein
folding, where hydrophobic regions tend to be on the interior of the folded
structure. Such exposure will promote aggregation or self-association,
possibly leading to physical instability and potential loss of biological
activity, because the interaction with the receptor site requires folded
structures with the correct conformation.
168. And then at page 71:
7.2. Aggregation
Protein aggregation is caused mainly by hydrophobic interactions
resulting from denaturation. When the interior hydrophobic region of a
partially or fully unfolded protein is exposed to water, this creates a
thermodynamically unfavorable situation due to the fact that a hydrophobic
interior is now exposed to a hydrophilic aqueous environment. Consequently,
the decrease in entropy from structuring water molecules around the
hydrophobic region generates a driving force for the denatured protein to
aggregate, mainly through the exposed hydrophobic regions. Thus, solubility
of the protein is also compromised (Franks, 1994). In some cases self association
of protein subunits, either native or misfolded, may occur under
certain conditions and this may lead to precipitation and loss in activity
(Brange and Langkjaer, 1993; Brange et at., 1 992a,b; Mitraki and King, 1989;
Shahrokh et at., 1994a; Silvestri et at., 1993).
169. It goes on to deal with surfactants at page 73 in the following terms:
8. FORMULATION APPROACHES FOR SOLVING
PHYSICAL STABILITY PROBLEMS
8.1. Surfactants
Surfactants, as their name implies, are surface-active agents, which can
exert their effect at solid-liquid, liquid-liquid, and liquid-air interfaces
because of their chemical composition, which includes both hydrophilic and
hydrophobic groups. These materials reduce the concentration of proteins in
dilute solutions at the surface of the solution where they can be adsorbed
and/or denatured. Surfactants can bind to hydrophobic interfaces in protein
formulations and packaging. Glass, rubber, or plastic adsorption of proteins
is well documented (Chawla et ai., 1985; Suelter and DeLuca, 1983).
Proteins on the surface of water will aggregate, particularly when shaken,
because of unfolding and subsequent aggregation of the protein monolayer.
Surfactants can denature proteins, but also can stabilize proteins against
surface denaturation.
Generally, ionic surfactants tend to denature proteins. Nonionic
surfactants usually do not denature proteins even at relatively high
concentrations (1 % w/v) (Cleland et ai., 1993). Most parenterally acceptable
nonionic surfactants come from either the polysorbate (sorbitol-polyethylene
oxide polymers) or polyether (polyethylene oxide-polyproplyene oxide block
co-polymers) groups. Polysorbates 20 and 80 are the only known surfactant
stabilizers in marketed protein formulations (see Table IV). However, other
surfactants used in protein formulations for clinical studies and/or found in
the patent literature include polysorbate 20, Pluronic F68, and other
polyoxyethylene ethers (e.g., the "Brij" class) (Wang and Hanson, 1988).
170. And then addresses silicone at pages 96-7:
11.4. Silicone
Silicone generally is required to lubricate the rubber closure to provide
surface lubricity such that closures or stoppers will not "clump" during
and after autoclaving and will "flow" in high-speed stoppering equipment.
Silicone coating may also facilitate the proper fit of closures into the necks
of vials, thus increasing the integrity of the container/closure interface.
Insufficient surface lubricity can cause "clumping" or "twinning" of stoppers
resulting in a shutdown of the filling line. Not only can this be costly, it can
also jeopardize assurance of sterility, because of human intervention on the
filling line to remove poorly siliconized or nonsiliconized stoppers. Glass
vials of suspension products sometimes may be siliconized to facilitate
product drainage and help to assure uniformity of the withdrawn dose.
Silicone also is used in cartridge delivery systems where a rubber plunger
must be pushed easily inside the glass barrel to accurately deliver the right
volume of solution and dose of protein. Quantities of silicone applied to
rubber closures are considered extremely low with respect to potential
chemical and biological incompatibilities of this relatively inert material
(Riffkin, 1968).
Silicone represents a hydrophobic surface that can cause problems with
some proteins. Glass surfaces coated with silicone, originally in the
expectation of decreasing protein adsorption to glass, in fact were found
to increase the adsorption of a variety of proteins, including insulin,
globulin, and lysozyme (Mizutani, 1981). Only albumin did not bind to the
siliconized glass surface as much as it did with nonsiliconized glass, an
observation leading to the use of albumin in minimizing adsorption of other
proteins to glass.
171. I have quoted the whole of this rather long passage to illustrate that there is more teaching about the issues with silicone than just hydrophobicity, but that issue is called out distinctly and would strike the skilled formulator, in my view.
172. Finally, there is a long section about stability from page 107. A key section is at 111:
13.4. Stability Studies Supporting Distribution of Protein Products
Formulation development typically focuses on protein dosage form
stability under well-controlled conditions. However, studies must be done to
demonstrate that the final protein formulation in the final package produced by
a validated process will retain its stability and other quality properties during
distribution of the product throughout the world. These studies can be
accomplished both by simulation in the laboratory and actual distribution of
the product. During distribution, even if the product is to be held in controlled
(e.g., refrigerated) conditions, aberrant situations can occur, such as due to
transportation breakdown, mechanical failure, dropping or otherwise mishandling
packages, and even overt violation of required handling procedures.
Data should be available to aid in knowing what these extreme situations will do
to the quality of the product. The effect of shear (i.e., agitation, mixing, and
other mechanical forces experienced during processing and handling) and
temperature extremes on protein stability during simulated and actual
conditions should be evaluated.
173. Wyeth’s approach to Akers was characteristically attritional (apart from the overriding point that therapeutic proteins were completely distinct from vaccines, which I have rejected). I found it unrealistic. For example, it submitted that the passage on pages 96-97 does not talk about aggregation. This is true in absolutely literal terms, but it refers to problems of hydrophobicity and adsorption and I am sure the skilled reader would have in mind what had already been said about hydrophobicity and aggregation at page 71.
174. I conclude that Akers supports MSD’s position on the issues of the relevance of packaging, hydrophobicity and aggregation, the use of surfactants to address such problems, silicone as a likely cause of such problems arising inter alia from hydrophobicity, and the general need for stability testing.
175. Bontempo is another textbook, from 1997, entitled “Development of Biopharmaceutical Parenteral Dosage Forms”. It is not about vaccines specifically and the parts relied on by MSD refer generally to proteins. It thus raises similar issues in this respect to Akers, and my conclusion is the same in the light of my finding that the skilled team would be aware of the situation with therapeutic proteins.
176. It is fair to say that Prof Crommelin overstated how much support for his views can be found in Bontempo, but I do not think this was deliberate or malicious.
177. Bontempo refers to shake tests at pages 99 and 121. It is not necessary to quote them, but they support MSD’s case that shake tests would be done early in development, and in the final form in due course.
178. Bontempo refers to aggregation at page 104. It says this:
C. Aggregation
Protein aggregation derived from either physical or chemical inactivation, is presently a major biopharmaceutical problem (17-21). Aggregation can be either covalent or noncovalent, occurring during any phase of product development from purification to formulation. An early detection of aggregation via biochemical or spectrophotometric methods, or both, can be of significant guidance to formulation scientists in selecting compatible excipients to minimize and/or prevent its formation in the experimental formulation.
Formation of aggregation can begin by the formation of initial particles from protein molecules via the Brownian movement. This is followed by collision of these molecules and aggregates of varying sizes can be formed. These aggregates can be generated by shear or collisional forces (22). Detection and measurements of aggregations can be performed by a number of techniques. [And it then lists some, including visual observation.]
179. This is of only moderate relevance; it does not discuss the mechanism by which aggregation occurs in detail, and what it does say (about Brownian movement) is, on its face, just part of the picture. It supports Dr Vanden Bossche’s view that hydrophobic interactions are not the only cause of aggregation, as I have noted above. But this is a side issue.
180. Bontempo discusses siliconization at page 120, where it says:
E. Siliconization of Elastomeric Closures
Siliconization of elastomeric closures, with a 2.0% solution of Dow Corning 360, was usually necessary to give an elastomeric closure better insertion into the neck of a glass vial. High speed filling certainly required this treatment. Without it, all kinds of problems arose during manufacturing. However, with proteins and peptides, significant problems were encountered in dealing with potential adsorptive problems between the protein-silicone elastomeric interactions (unpublished data). Silicone traces also interfered with the development of analytical methodology, for it complexed readily with the proteins.
181. This identifies in very general terms that there have been numerous adsorptive problems with silicone. It supports MSD’s case that the skilled person would regard silicone as a potential source of problems, and the description of them is not inconsistent with MSD’s case, but it contains no real explanation of the mechanism and no pointer towards surfactants.
182. Overall, Bontempo is significantly supportive of MSD’s case on shake tests and a generally cautious approach to silicone, but otherwise takes matters no further.
183. Powell and Newman is a 1995 book called “Vaccine Design - The Subunit and Adjuvant Approach”. It is therefore squarely of relevance to the vaccine field and not open to the criticism that Wyeth levelled at the therapeutic protein materials.
184. Instead, Wyeth argued that the book received little focus in Prof Crommelin’s first report. This is true and I have taken it into account, but I found it of little help. The book looks very much like the sort of level of detail and of practical utility that would be CGK.
185. Powell and Newman identify that a vaccine should be stable to agitation (foot of page 23), although this was not in dispute in those general terms.
186. It is also of relevance to a modest extent in showing that the skilled person would have regard to non-vaccine sources: at page 24 second full paragraph it refers the reader in relation to further understanding of degradation pathways to a number of more general (non-vaccine) publications. It was not said that those specific cross-references were relevant to the issues I have to decide, but they show an openness to referring to non-vaccine sources and to that extent provide some support to MSD’s case generally.
187. The key passages of Powell and Newman, however, were on page 25 in relation to adsorption and the use of surfactants. The authors write as follows:
“ Physical instability of proteins that are used as immunogens may compromise vaccine formulation stability. This can occur in several ways such as loss of protein via adsorption of the protein immunogen to the surface of the container or possibly by protein aggregation and subsequent denaturation. Surface adsorption usually occurs at low concentrations of protein and in the absence of carriers. The adsorption of protein has more profound implications than just the simple loss of a component in the formulation because adsorption increases protein susceptibility to denaturation (Steadman et at., 1992). A well-characterized example is insulin which readily adsorbs to hydrophobic surfaces and subsequently denatures. Denatured insulin molecules then accumulate resulting in the formation of nonfunctional aggregates (Sluzky et al., 1991; Sato et al., 1984). Other effects associated with protein adsorption and aggregation have been extensively reviewed (Kiefhaber et al., 1991; Norde and Lyklema, 1991; Andrade et al., 1992; Sadana, 1992).
Protein adsorption can be prevented by increasing the protein concentration in the formulations or by the addition of appropriate carrier molecules, such as particulate alum or surfactants. Surfactants, such as those used in many emulsion-based adjuvant formulations, are useful because they bind to the hydrophobic areas of both the soluble protein and the container surfaces and inhibit protein adsorption. Examples of these types of compounds are the block copolymers and the polysorbate polymers, both of which have been used in vaccine formulations as adjuvants. The polysorbate polymers consist of a sorbitol-polyethylene oxide head group and a hydrocarbon tail, whereas the block copolymers consist of a polyethylene oxide-polypropylene oxide copolymer.”
188. To my mind, this provides clear teaching about the problem of adsorption and aggregation, and that surfactants are useful to address the problems because of their binding to the hydrophobic areas of the protein and the container surfaces, to prevent adsorption. It is not a long section of text but that does not detract from what it is saying, and it does not need to be long because the skilled formulator would find it easy to understand.
189. Wyeth’s main answer to this teaching was a semantic analysis of the first sentence of the second paragraph, and in particular “the addition of appropriate carrier molecules such as particulate alum or surfactants”. Wyeth said that the way this was written meant that the authors were only talking about surfactants when used as carriers. In a sense I think its point was that had the authors been referring to surfactants other than as carriers there would have been an extra comma after “alum”, to decouple the surfactants from the qualifier “carrier”, thus “the addition of appropriate carrier molecules such as particulate alum, or surfactants”. I thought this was much too pedantic and in the overall context I preferred MSD’s broader reading that the authors were referring to surfactants in general, but it really does not matter because the next sentence, which Wyeth largely ignored, absolutely spells out that surfactants may be used to address adsorption (and therefore aggregation).
190. Powell and Newman do not refer expressly to silicone, but rather to “surfaces”, but there is much other material in the other documents I consider to identify that silicone provides a relevant interface at which adsorption can take place.
191. Overall, Powell and Newman provide powerful support for the use of a surfactant being a practical solution for a problem of adsorption or aggregation of a vaccine at an interface where hydrophobic regions of a vaccine begin to be exposed.
192. MSD relied on a number of trade publications from, or (friendly) interviews with, syringe manufacturers. There can have been no suggestion that the materials themselves were CGK. If there was, I reject it. But I understood them to be advanced to show that the issues under discussion were widely canvassed, and I agree with the notion that if the syringe manufacturers were publicising this kind of issue they were also discussing it with their customers, the skilled formulators.
193. However, one must scrutinise the content of such materials carefully. All they really established was that the manufacturers were aware, no doubt from feedback from customers, that too much silicone caused problems because it could interact with the contents of the syringe.
194. This provides strong additional support for it being CGK that silicone generally was a “red flag”. But it does not support it being CGK that the problems could arise specifically from aggregation owing to hydrophobic interactions.
195. One of the syringe publications (DJAC 10) did specifically mention aggregation and the presence of particulates, but it was much too general to be of help to MSD, and was a lone instance.
196. A similar but more cogent piece of evidence advanced by MSD was a 2004 article by Amy Rosenberg of the CDER division of the FDA. Its title was “A Risk-Based Approach to Immunogenicity Concerns of Therapeutic Protein Products”.
197. At page 204 it said this:
Container-closure considerations.
FDA has come to increasingly appreciate the fact that interactions between
protein therapeutics and container closures may negatively
impact product quality and immunogenicity. This is particularly apparent
with the increasing use of prefilled syringes for patient self-administration.
Syringes are delivery devices with multiple surfaces and materials that can
interact with the product and alter product quality. Glass and air interfaces
are hydrophobic surfaces that can mediate protein denaturation and aggregation.
Moreover, glass syringes and movable surfaces (such as syringe plungers)
are coated with silicone oil, which facilitates injection but also provides a surface
on which proteins can denature and aggregate.
198. As with the syringe manufacturers’ materials, this document was not itself CGK, but it shows that the FDA was very much aware of silicone as a potential problem by the mechanism of hydrophobic surfaces mediate protein denaturation and aggregation. Plainly, the FDA’s growing awareness must have involved regular interactions with skilled formulators. So it supports MSD’s case that silicone as a cause of aggregation was CGK.
199. Wyeth objected that this document was not concerned with vaccines, and, relatedly, that the CDER division of the FDA was not tasked with vaccines. I have dealt with the former point multiple times already and the latter was merely an aspect of the administrative arrangement of the FDA.
200. When Wyeth encountered the problem with aggregation it went to the literature to seek information. One of the documents it found was Jones, “Silicone Oil Induced Aggregation of Proteins”, published in the Journal of Pharmaceutical Sciences in April 2005. Prof Crommelin suggested that this paper would also be found by the skilled formulator conducting a literature search if seeking to address a problem of aggregation encountered in the presence of silicone.
201. I was not satisfied that MSD proved that Jones would necessarily be found in this way, and it was not alleged to be CGK in itself. The evidence was lacking as to what search would be done, or with what goal, and nor was it shown how much material in addition to Jones might be thrown up.
202. In the light of my conclusion that Jones would not necessarily be found, there is no need to go into the complex argument that developed over exactly what it shows, or its experimental design or results.
203. However, Jones is relevant in another way, which is that the introductory paragraphs contain the following statements:
“Silicone oil contamination has long been suspected of being responsible in some cases for the aggregation seen in certain protein pharmaceutical preparations. Several publications in the 1980s implicated the release of silicone oil from disposable plastic syringes in the aggregation of insulin.”
204. And:
“In addition, it is the author’s experience that questions of silicone oil contamination and its potential role in protein aggregation arise frequently during the pharmaceutical development of proteins generally, although little information about this potential problem is available in the scientific literature.”
205. Both these provide further support for it being CGK that silicone oil was a likely common cause for protein aggregation.
206. I have already referred to Dr Khandke’s evidence about the January 2005 Wyeth document mentioning silicone. I have rejected her evidence that that was not the focus of the document. It shows that Wyeth had had numerous problems with silicone, including aggregation. The documents which emerged after trial appear to reinforce Wyeth’s having had such problems prior to 2005 and prior to the project which led to the Patent.
207. Wyeth is only one company and I must be careful not to assume that the inventors’ organisation was necessarily representative of the workplace of the notional, ordinary skilled formulator. But there were only a few companies in the vaccines field and the fact that one of them had had such a number of issues with silicone supports MSD’s case.
208. Chiron is the cited prior art. In itself it clearly was not CGK and MSD does not submit that it was. As appears below, it refers to the inclusion of a surfactant (Tween) “to minimise adsorption of antigens to containers”. This shows that another company in this sparsely-populated field was aware of the issue of aggregation with vaccines (though silicone is not specifically mentioned) and that the solution might be a surfactant. There is no suggestion that the information is surprising, or needs explanation, or that it is inventive (there is no claim to it).
209. This provides further, albeit modest support for MSD’s case.
210. In support of its case that it would be obvious to use a surfactant in this kind of vaccine to deal with aggregation, MSD relied on a number of specific commercial vaccines.
211. To assess this, it is necessary to identify what is at issue.
212. First, the question relevant to my decision is whether CGK use of surfactants in actual vaccines would support the idea of using them to deal with aggregation, not whether it would be acceptable to have a surfactant in this type of vaccine at all. I mention this because at an early stage in the trial I thought Wyeth might be going to say that there would be perceived safety issues with surfactants. Had that been the case, it could have been relevant for MSD to identify any vaccine that contained a surfactant, for whatever reason. But Mr Tappin disavowed any such argument.
213. Second, the mere presence of a surfactant in a vaccine of this type would not necessarily imply anything about whether it was being used to deal with aggregation (silicone-induced or otherwise). This is because, as I explain above, surfactants have many uses, including, for example, lysing cells to release proteins. As a result, the presence of a surfactant in a vaccine could be because it was added at the final stage of formulation and so could be a deliberate excipient to address aggregation, or it could be because it was a residue from the process by which the antigens were produced in bulk at an earlier stage. It might be possible to infer which was the case from the quantities given, or from the public literature concerning a given vaccine, whether a regulatory filing or otherwise.
214. Prof. Crommelin originally identified 12 approved vaccines said to contain surfactant. Of these, 9 were later accepted to contain merely residual surfactant, not excipients added at the formulation stage. Three remained.
215. The first was VAXEM HIB, a vaccine from Chiron. Neither expert had heard of it at the priority date. Prof Crommelin said it would have been found from a literature search. It was a matter of dispute between the experts whether the materials that would be found showed that the TWEEN 80 that was in it was added at the formulation stage. Prof Crommelin accepted that the details given in the documents were inconsistent.
216. The second was GARDASIL, a Merck product published after the priority date of the Patent. Again, it was disputed whether this was added as an excipient, or merely residual.
217. The third was HAVRIX (and there was an earlier related product called TWINRIX), an inactivated Hepatitis A virus formulation. Prof Crommelin accepted that it was possible that the TWEEN 20 used in it was added to lyse the cells or solubilise the proteins at the bulk stage. Prof Crommelin again accepted that the details were inconsistent.
218. In addition to those three, MSD relied on five candidate (i.e. not yet approved) vaccines. They were two Wyeth vaccines in phase I, an MSD adenovirus vaccine, a vaccine called StaphVAX based on a 2002 paper from Shinefield and a nicotine vaccine for smokers called NicVAX.
219. I found it difficult to follow, during the trial, which of these vaccines were said by MSD to be CGK, which were said to evidence CGK, and which would be found on a literature search. I directed MSD to make a table identifying these matters, and for Wyeth then to comment on it. This was done.
220. I was grateful for the provision of the table, which showed that the issues could be grouped together to some extent. In particular, it revealed that the candidate vaccines were not said to be CGK themselves, merely reflective of CGK or such as to be found on a literature search. It confirmed that the vaccines said actually to be CGK and to have included a surfactant as an excipient were GARDASIL and HAVRIX (VAXEM HIB was demoted to reflective of the CGK or the literature search category). I can trim this down further since I accept Wyeth’s submission that GARDASIL could not be CGK because it was not approved until later.
221. One swallow does not make a summer. HAVRIX is far too slight as a piece of evidence to establish that the use of surfactants as excipients in these vaccines actually was CGK (category 1 in the table), and even if it were well known as a product that existed, there are real doubts about whether the surfactant was included as an excipient or not, and I do not accept that its specific excipients were CGK in any event.
222. I deal next with the allegation that a large number of vaccines would have been found on a literature search (category 3 in the table). I agree with Wyeth that proving this would require concrete evidence of the search to be done, its goals and search terms, and specifically what would be found. It is not good enough to put a lot of materials in evidence and then make general allegation that they could be found on a literature search. Perhaps they could, but it is still necessary for a party making this kind of attack to show what would be found by obvious means, and what would be done with it. Requiring any less would be unfair on patentees because it could in practical terms allow the elevation to CGK of any published material.
223. Last, I address the materials said to reflect CGK (category 2 in the table - I have taken it out of order). The task with this category is to determine what it might add. I have held on the basis of the textbooks and other materials that I discuss above that the use of surfactants to reduce silicone-induced aggregation was CGK based on the evidence of Prof Crommelin, the textbooks, regulatory and trade materials, and so on. If I had not done so, materials said to be “reflective” of common general knowledge could not redress MSD’s failure. But given that I have, what need is there for further “reflective” materials? Overall, I find some modest though unnecessary support for my conclusions in the fact that some vaccines actually did have surfactants in them and (reverting somewhat to category 3) I think it reasonable to say that a formulator who already had it in mind to use a surfactant from other CGK sources (as I find is the case) might do a literature search for vaccine product labels to gain confidence that he or she was not doing something unprecedented (though I do not think they would find that likely), and would thereupon probably find some evidence of surfactant use. It cannot go further than that, and is not necessary to my conclusion.
224. So in the main I reject MSD’s case based on the real vaccines. This does not mean that vaccine companies were not using surfactants to prevent aggregation. They very well may have been. It just means that analysis of public information about those vaccines would not inform the skilled formulator about whether or not they were.
225. Some further specific materials were relevant to whether stability testing, and if so of what kind was CGK.
226. MSD’s position was that it was CGK to test vaccine formulations to see that they would remain stable, including physically stable, during (in particular) shipping and storage over its expected shelf life. MSD’s case focused on testing physical stability against shear forces and agitation.
227. MSD also emphasised the performance of tests early in a product’s development.
228. To make this part of its case MSD relied on:
a) The textbooks: Akers, Bontempo and Powell & Newman which I have covered above;
b) Various recommendations of the ICH;
c) The experience of Wyeth as illustrated by Dr Khandke;
d) The experience of Prof Crommelin and his evidence generally.
229. Akers and Bontempo were accepted by Wyeth to refer to agitation tests for protein drugs (and they refer to processing, handling and stability in the final container), but Wyeth again said that that would not be relevant to vaccines. I reject that argument for the same reasons as above. Powell & Newman refers to the need for vaccines to be stable against agitation but Wyeth submitted that it did not teach the need to test for such stability. That is unrealistic in my view, although it is fair to say that the book is not specific about what tests should be done or when.
230. The ICH guidelines were ICH Q1A and ICH Q5C. They were, to my mind, surprisingly general and one might even say vague. They do not refer to agitation tests specifically but they do refer to transportation, as to which I accept MSD’s case that the skilled formulator would appreciate it was an occasion when agitation and shear forces would be experienced.
231. Wyeth did real time and stressed tests for agitation at an early stage. This supports MSD’s case, as does Dr Khandke’s evidence that tests were needed to satisfy the regulator and would at some point have to be done using the final container. At a later stage, Wyeth also did a test called the ISTA test.
232. Prof Crommelin was firm that agitation tests were standard, albeit that the documentary sources he referred to were chipped away at somewhat. Also, his evidence on this topic was hindered by the attitude to confidentiality to which I have referred above.
233. Dr Vanden Bossche’s evidence on this topic was extreme. He said that the only test done would be a visual inspection in the final container, late in the development process. He said that the skilled formulator would not consider there to be a need to test with aluminium adjuvanted vaccines because the skilled person would have unwavering confidence that adsorption to the adjuvant would prevent aggregation, and (relatedly) that the skilled formulator would test for adsorption to the adjuvant and not directly for any effect on its stability during simulated shipping or the like. To my mind none of this made any practical sense, did not fit with the real world evidence of Dr Khandke at Wyeth and was not supported by any document.
234. There are certainly weaknesses in MSD’s evidence on this issue but taking all the above matters as a whole I hold that it was CGK to test for stability in terms of resistance to aggregation at an early stage of development, and in the intended final container. The tests would seek to replicate or simulate the real world effects expected, including shear forces and agitation.
235. MSD’s case is that the skilled team would only consider adding a surfactant if there was evidence of a problem that needed addressing; the team would not consider adding a surfactant just prophylactically, in case of adsorption or aggregation. So the question arises of whether the skilled team would notice aggregation, if it was happening.
236. Wyeth did not accept that that the skilled team would notice aggregation. It actively argued that it was not part of the CGK to test for aggregation routinely, and it also (though more passively, I sensed), took issue with whether the skilled team would spot aggregation even if it were looked for.
237. I have resolved the first point against Wyeth and held that it was CGK to test for aggregation. I have held that there was no specific test required by the regulators, but that the skilled team would carry out tests during product development to simulate the conditions that the vaccine would encounter in shipping, storage and use.
238. Even if I were wrong about that and the skilled team would not conduct such tests at an early stage, aggregation could of course occur in due course when the product was in fact shipped, stored and used.
239. The second aspect - whether aggregation would be noticed if and when it were to happen with this kind of vaccine - involves consideration of what happened at Wyeth, and I deal with that in a moment. It occupied a lot of time at trial. However, this is the sort of situation where it is important to put the invention story in its proper place. Wyeth represents just one skilled team working in a particular set of circumstances, and the question for me is what the notional skilled team would do without invention, based on its CGK. As it happens, I think the Wyeth story supports MSD’s case, but even if it had helped Wyeth I would still have needed to consider the notional skilled team.
240. Prof Crommelin’s evidence was that aggregation could and would be identified by routine means - visual inspection or testing for loss of antigenicity in particular, and I accept that evidence, which I think is the primary evidence on this issue.
241. I turn to the Wyeth story. The detail in which this was explored at trial was unnecessary in my view, and anyway the only witness that I heard from was of course Dr Khandke, and many others were involved. I think I can, however, draw conclusions about the key events.
242. The first observation of aggregation in siliconized containers did not happen with a formulation with an aluminium adjuvant, but with a different formulation intended for adults and which had no aluminium adjuvant. Aggregation with that formulation was noticed after a shake test, and that led Wyeth to go back and review the formulation that did have an aluminium adjuvant (intended for infants). Upon doing so, aggregation was spotted, but it was harder to see. It seems therefore that it was probably overlooked at an earlier stage.
243. The Patent at [0087] says that aggregation was harder to see in the infant formulation including the aluminium adjuvant. Dr Khandke’s evidence was that it could be seen by “[t]hose with a trained eye”.
244. The fact that it was possible to miss the aggregation (and that it was missed initially) does not assist Wyeth in my view. The Patent and Dr Khandke both support the conclusion, which I accept, that although the adjuvant made it harder, with care and a trained eye aggregation would be noticed. Carefulness and a trained eye are exactly the characteristics of the ordinary skilled team. They would notice the aggregation without the need for any invention (or luck). I do not know quite how the workers at Wyeth who missed the aggregation earlier on did so, but it does not matter. For whatever reason they did not meet the standards of the notional skilled team.
245. I should mention two other points on this issue.
246. The first is a forensic point by MSD: how, it asked, could it be an invention to resolve a problem which was so slight that it was not even noticeable at all? Mr Tappin avoided engaging with this, perhaps appreciating that it would be necessary either to accept that the aggregation was bad enough to notice, or that it was indeed so slight that it was not a real problem at all. I thought it was a persuasive point but it is not necessary to my reasoning, and ultimately it was MSD’s burden as part of its obviousness case to prove that the problem would be detected without the need for invention, which it did for the reasons set out above.
247. The second point concerned the amount of silicone used and its impact on aggregation. It seems likely that the amount of silicone in the various batches of containers used at Wyeth was variable (with the variation being random), and it is possible that this accounted for when aggregation was observed and when it was not. I thought the evidence on this was inadequate to draw any firm conclusion, and there is no reason to think that there was the need for any unusually high amount of silicone to be present for aggregation to occur. So the point does not affect my finding that aggregation would be detected by routine means and without invention.
248. Dr Vanden Bossche did not really oppose the proposition that if aggregation were to be observed it would be straightforward to identify silicone as the cause. In particular he accepted that it would be attributed to the presence of silicone in the event (as happened at Wyeth and as would be likely in ordinary development by the notional skilled team) that the aggregation was first seen after the vaccine was first kept in siliconized containers.
249. The claims of the Patent in relation to which I potentially have to make decisions given the narrowing of issues that I have mentioned are as follows (taken from MSD’s opening written skeleton and including the upper case reference letters inserted by MSD and to which I refer below in dealing with claim construction):
Claim 1
A. 1. A siliconized container means filled with a formulation which inhibits silicone induced aggregation of a polysaccharide-protein conjugate comprised in a siliconized container means, the formulation comprising
B. (i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5,
C. (ii) an aluminum salt and
D. (iii) one or more polysaccharide-protein conjugates
E. wherein the polysaccharide-protein conjugate comprises one or more pneumococcal polysaccharides
F. and wherein the one or more pneumococcal polysaccharides are a S. pneumoniae serotype 4 polysaccharide, a S. pneumoniae serotype 6B polysaccharide, a S. pneumoniae serotype 9V polysaccharide, a S. pneumoniae serotype 14 polysaccharide, a S. pneumoniae serotype 18C polysaccharide, a S. pneumoniae serotype 19F polysaccharide, a S. pneumoniae serotype 23F polysaccharide, a S. pneumoniae serotype 1 polysaccharide, a S. pneumoniae serotype 3 polysaccharide, a S. pneumoniae serotype 5 polysaccharide, a S. pneumoniae serotype 6A polysaccharide, a S. pneumoniae serotype 7F polysaccharide and a S. pneumoniae serotype 19A polysaccharide.
Claim 2
2. The siliconized container means of claim 1, wherein the formulation further comprises a surfactant.
Claim 4
4. The siliconized container means of claim 1, wherein the formulation further comprises polysorbate 80 (TweenTM 80).
Claim 16
16. Use of a formulation to inhibit silicone induced aggregation of a polysaccharide-protein conjugate comprised in a siliconized container means, the formulation comprising
(i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5,
(ii) an aluminum salt, and
(iii) one or more polysaccharide-protein conjugates
wherein the polysaccharide-protein conjugate comprises one or more pneumococcal polysaccharides
wherein the one or more pneumococcal polysaccharides are a S. pneumoniae serotype 4 polysaccharide, a S. pneumoniae serotype 6B polysaccharide, a S. pneumoniae serotype 9V polysaccharide, a S. pneumoniae serotype 14 polysaccharide, a S. pneumoniae serotype 18C polysaccharide, a S. pneumoniae serotype 19F polysaccharide, a S. pneumoniae serotype 23F polysaccharide, a S. pneumoniae serotype 1 polysaccharide, a S. pneumoniae serotype 3 polysaccharide, a S. pneumoniae serotype 5 polysaccharide, a S. pneumoniae
serotype 6A polysaccharide, a S. pneumoniae serotype 7F polysaccharide and a S. pneumoniae serotype 19A polysaccharide.
Claim 17
17. The use of claim 16, wherein the formulation further comprises a surfactant.
Claim 19
19. The use of claim 16, wherein the formulation further comprises polysorbate 80 (TweenTM80).
250. I have omitted claims which are relevant to interpretation but on which there is not a specific issue (in particular, claim 13).
251. The central issue is whether integer F of claim 1 requires precisely the 13 identified S. pneumoniae serotype polysaccharides, or requires those 13 but permits more. This hinges on the word “are” in integer F, although of course the claim as a whole must be construed in context.
252. Conventionally in patent claim drafting the word “comprises” is used to indicate “includes”, meaning that those things that follow must be present, but others are allowed in addition.
253. In claim 1, “comprised” or “comprises” is used several times.
254. In integer A the polysaccharide-protein conjugate is first required to be “comprised in” a siliconized container means. This merely means “contained in” and is not relevant to the disputed point of construction that I am dealing with; I mention it only for completeness.
255. Then, integer A also stipulates “the formulation comprising”, followed by three components numbered (i) to (iii). So all three must be present, but other ingredients are also allowed, which would permit the inclusion of any other excipient, such as a preservative. The skilled reader would understand this, and understand that it would not make sense for the patentee to exclude from the claim some additional excipient that would not affect the invention.
256. Integer E requires that the third of the three ingredients, (iii), which is “one or more polysaccharide-protein conjugates” “comprises one or more pneumococcal polysaccharides”.
257. This has the clear effect that the formulation must contain pneumococcal polysaccharides and may contain others, such as meningococcal or streptococcal polysaccharides. It was common general knowledge that vaccines might contain more than one type of polysaccharides in this way, and there is basis for it in the specification at [0015].
258. Thus far, therefore, the language is inclusive rather than limiting.
259. Integer F, however, does not use the word “comprises”, but the word “are”. The contrast is obvious and powerful, and the ordinary meaning of “are” does not extend to “include”. I consider it striking that if the patentee had meant that other pneumococcal polysaccharides could be included in addition to the 13 listed, it would just have used the word “comprise” again.
260. Wyeth submitted in its written opening that:
a) The effect of the word “comprising” in feature A (referred to by Wyeth as “(b)” and misquoted as “comprises” in paragraph 192 of its opening) was that other polysaccharides were also allowed.
b) The effect of the word “comprises” in feature E (referred to by Wyeth as “(c)”), taken with the opening words of A “a formulation … .conjugate” (referred to by Wyeth as “(a)”) was to avoid any limitation to the 13 identified serotypes.
261. I reject both these points.
262. The first use of “comprising” in feature A requires the three specified things in B to D to be present, and allows other things too. But feature D just says as a generality that some polysaccharide-protein conjugates have to be present and the reader would see that more about them is explained in features E and F. By focusing on feature A Wyeth avoids focusing on whether or not feature F is limiting.
263. As I have explained above, the use in feature E of “comprises” allows other bacterial polysaccharides but requires that one or more is pneumococcal. Wyeth’s taking it in combination with feature A/(a) takes this no further other than, again, to avoid dealing with feature F, whose function I have already said I consider to be to specify that the set of pneumococcal polysaccharides required must be (“are”) exactly the 13 listed.
264. It is hard to see how else, realistically, following conventional drafting, the patentee would have phrased the claim if it had intended to specify precisely the 13 pneumococcal saccharides.
265. So far I have focused on the words of the claim itself but I have also taken into account the textual and scientific context.
266. Textually, Wyeth relied on claim 13. I thought originally (as I think did MSD) that Wyeth was saying that claim 13 would be redundant unless claim 1 were interpreted as it contended. That would have been wrong since at the very least claim 13 requires that the polysaccharide be conjugated to CRM197, which is not required in claim 1.
267. However, in fact Wyeth’s point was simpler, which was that when the patentee wanted to refer to a 13v pneumococcal formulation it had the expression to hand, namely “13vPnC”. It pointed to paragraphs [0014], [0024] and [0061] as well.
268. I found this unconvincing since the availability of other language that could have been used does not detract from the clear words that actually are used in claim 1, and in any event paragraphs [0014], [0024] and [0061] all use the word “comprising” while claim 13 does not: if the patentee had intended to lift permissive wording from the specification it would have used the wording of those paragraphs and left in the “comprising”.
269. From a scientific point of view, a set of serotypes is selected as a careful and precise combination balancing efficacy, cross-protection and ability to be manufactured. Adding serotypes could risk the overall package, in particular prejudicing manufacturing feasibility for perhaps inadequate compensating gain in efficacy or cross-protection. Adding serotypes is not something that the skilled person would think could be done lightly or arbitrarily - indeed that is part of Wyeth’s defence to obviousness - so there is a real rationale for the patentee to limit the claims to the precise combination of serotypes, which could be formulated (as the Patent shows) and is taught by the Patent (though not proved) to be useful clinically.
270. Taking all these considerations into account, I reject Wyeth’s submission and accept MSD’s. On that basis, there is no infringement by v114 of claim 1 or dependent claims. It was common ground that whatever conclusion was reached on claim 1 also applied to claim 16 and dependent claims.
271. As I have explained above, this would only matter in the event of my finding that claims 1 and 16 are infringed (but anticipated), that the obviousness attacks fail, and that claims 2 and 17 (to surfactants generally) would be invalid for added matter.
272. I have found that these things do not apply: claims 1 and 16 are not infringed because MSD’s product is a 15v vaccine, and claims 2 and 17 are not bad for added matter (see below).
273. In addition, I have found all the claims invalid for obviousness over de la Pena, but claim 16 novel over Hausdorff 381 (again, see below).
274. So for multiple cumulative reasons the equivalence argument is irrelevant to my overall conclusions of non-infringement and invalidity.
275. In addition, MSD accepted that the first and second Actavis questions were to be answered in Wyeth’s favour. So in the event that this action were to go on appeal and my other findings reversed so as to make equivalence relevant, the Court of Appeal would have to decide an issue of pure interpretation of the claims in the context of the specification.
276. In other words, there are no findings of fact that I need to make to equip the Court of Appeal to decide the issue if it ever were to become relevant. It is a point of some complexity, and likely to involve consideration of the potential further development of this area of the law. This would be better considered in circumstances where it really matters.
277. For all these reasons I decline to decide the point.
278. The significance of the added matter case pursued at trial was to seek to prevent Wyeth from adding by amendment the limitations in claims 2 and 17 to a surfactant generally.
279. I did not understand there to be any dispute about the law, although the parties cited slightly different authorities for the relevant propositions.
280. Thus:
a) The general test for added matter is “whether a skilled man would, upon looking at the amended specification, learn anything about the invention which he could not learn from the unamended specification.” See the Court of Appeal in Nokia v. IPCom [2012] EWCA Civ 567 at [4]-[9], and see also at [56]-[60] stressing that the standard is whether the relevant information is directly and unambiguously disclosed in the unamended specification.
b) In relation to intermediate generalisation, the key principle (again, Nokia v. IPCom, ibid.) is that “it is not permissible to introduce into a claim a feature taken from a specific embodiment unless the skilled person would understand that the other features of the embodiment are not necessary to carry out the claimed invention. Put another way, it must be apparent to the skilled person that the selected feature is generally applicable to the claimed invention absent the other features of that embodiment.”
281. MSD also relied on the judgment of Arnold J in Conversant v. Huawei [2019] EWHC 1687 (Pat), but that simply summarises the ground covered by the Court of Appeal in Nokia v. IPCom (albeit in a helpful and pithy way) and does not change the picture.
282. Two allegations of added matter were pleaded. Only one was pursued, and it was as follows (Statement of Opposition, paragraph 6(a)):
“(a) The application as filed and the parent application each disclose a series of embodiments under the hearing “Summary of the Invention” from pages 3 - 12 of the parent application and in paras [0008] to [0050] of the application for the Patent as filed. The embodiment that is the subject of the claims of the Patent as proposed to be amended, namely that in which the formulation comprises:
(i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5;
(ii) an aluminium salt; and
(iii) one or more polysaccharide protein conjugates
is disclosed on p 7-9 of the parent application (paras [0022]-[0032] of the application for the Patent as filed). The only surfactant that is disclosed to be used with this formulation is polysorbate 80 (Tween 80) on p7 lines 27-30 (para [0025] of the application for the Patent as filed). Furthermore, the only example that relates to a formulation comprising an aluminium salt (Example 4) does not disclose the use of a surfactant. The only passages in the application as filed or the parent application which disclose a wider list of surfactants (e.g. p4, p6 of the parent application; paras [0011] and [0019] of the application for the Patent as filed), relate to different embodiments from that which is the subject of the claims of the Patent as proposed to be amended. Accordingly, claims 2, 3, 17 & 18 constitute an impermissible intermediate generalisation and accordingly added matter.”
283. The central thrust of this is that the amendment generalises the permitted surfactant from polysorbate 80 out to any surfactant, making the supporting point that where surfactants are disclosed more generally, other features of the claims are not disclosed. In other words, there is no disclosure of surfactants in general in combination with all the other claim features.
284. The parties drew my attention to the following parts of the application.
285. Page 7 lines 6-15:
In another embodiment, the invention is directed to formulations which inhibit silicone induced precipitation of a polysaccharide-protein conjugate comprised in a siliconized container means, the formulation comprising (i) a pH buffered saline solution, wherein the buffer has a pKa of about 3.5 to about 7.5, (ii) an aluminum salt and (iii) one or more polysaccharide-protein conjugates. In certain embodiments, the siliconized container means is selected from one or more of the group consisting of a vial, a vial stopper, a vial closure, a glass closure, a rubber closure, a plastic closure, a syringe, a syringe stopper, a syringe plunger, a flask, a beaker, a graduated cylinder, a fermentor, a bioreactor, tubing, a pipe, a bag, a jar, an ampoule, a cartridge and a disposable pen.
286. This refers to the problem of silicone induced aggregation (page 16 lines 25-29 make clear that aggregation and precipitation are used interchangeably) but not to the use of a surfactant.
287. Page 7 lines 27-30:
In certain other embodiments, the formulation further comprises polysorbate 80 (TweenTM80). In one specific embodiment, the final concentration of the polysorbate 80 in formulation is at least 0.01% to 10% polysorbate 80 weight/volume of the formulation.
288. This therefore refers to the “further” use of polysorbate 80 specifically.
289. Page 13 lines 5-14:
The present invention addresses an ongoing need in the art to improve the stability of immunogenic compositions such as polysaccharide-protein conjugates and protein immunogens. Thus, the present invention broadly relates to novel surfactant formulations and/or novel aluminum salt formulations which stabilize and inhibit precipitation of immunogenic compositions. More particularly, the invention described hereinafter, addresses a need in the art for formulations which stabilize and inhibit particulate formation (e.g., aggregation, precipitation) of immunogenic compositions which are processed, developed, formulated, manufactured and/or stored in container means such as fermentors, bioreactors, vials, flasks, bags, syringes, rubber stoppers, tubing and the like.
290. This refers in very general terms to the use of surfactants and/or aluminum salts for inhibiting precipitation.
291. Page 13 lines 32-35:
As set forth in detail herein, the present invention relates to the unexpected and surprising results that formulating an immunogenic composition with a surfactant such as TweenTM80 significantly enhances the stability and inhibits precipitation of an immunogenic composition.
292. This refers to the use of a surfactant “such as” TweenTM 80 to inhibit precipitation, but does not specifically mention silicone-induced aggregation (precipitation).
293. There is a further reference to silicone oil interactions at page 15 line 23, but not much weight was attached to this by either party, and in any event it is substantially repeated with significant additions at page 17 lines 5 to 11 under the heading “A. SURFACTANTS”:
As set forth above, the invention is directed to formulations which stabilize and inhibit aggregation of immunogenic compositions against the various factors which influence the stability of immunogenic compositions (e.g., shear forces, shipping agitation, silicone oil interactions, adsorption, manufacturing processes, temperature, humidity, length of time between manufacture and usage, etc.). In certain embodiments, the invention is directed to formulations comprising a surfactant.
294. And the possible surfactants for use are then described at lines 18-31 on the same page:
A surfactant used in a formulation of the present invention comprises any surfactant or any combination of surfactants which stabilizes and inhibits aggregation of an immunogenic composition described herein. Thus, a surfactant of the invention includes, but is not limited to, polysorbate 20 (TweenTM20), polysorbate 40 (TweenTM40), polysorbate 60 (TweenTM60), polysorbate 65 (TweenTM65), polysorbate 80 (TweenTM80), polysorbate 85 (TweenTM85), TritonTM N-101, TritonTM X-100, oxtoxynol 40, nonoxynol-9, triethanolamine, triethanolamine polypeptide oleate, polyoxyethylene-660 hydroxystearate (PEG-15, Solutol H15), polyoxyethylene-35-ricinoleate (Cremophor ELTM), soy lecithin, poloxamer, hexadecylamine, octadecylamine, octadecyl amino acid esters, lysolecithin, dimethyl-dioctadecylammonium bromide, methoxyhexadecylgylcerol, pluronic polyols, polyamines (e.g., pyran, dextransulfate, poly IC, carbopol), peptides (e.g., muramyl peptide and dipeptide, dimethylglycine, tuftsin), oil emulsions, mineral gels (e.g., aluminum phosphate) and immune stimulating complexes (ISCOMS).
295. But again, this paragraph in itself does not specifically refer to silicone induced aggregation.
296. It is worth saying that these passages do not come from the Examples of the specification (which in patent specifications generally are typically very specific and tend to be more self-contained). They come from the Summary of the Invention section and the Detailed Description of the Invention section. I do not suggest that there is any strict rule differentiating sections of specifications in terms of added matter, but the reader may well, depending on the context, expect that the more general sections prior to the Examples, will contain principles that may be more widely applicable than the individual details of the experiments in the Examples. I think this is reflected in the references in cases such as Nokia v. IPCom to taking features from specific embodiments out of context. Nonetheless, it could be added matter to combine parts of the general teaching in a way not taught in the application - it depends on the facts.
297. MSD is right that there is no specific disclosure in one paragraph in the application of the combination of all the features of claim 1 (or 16) with a surfactant generally.
298. However, that is not the end of the matter.
299. I consider that MSD’s attack takes the parts of the application referred to above too atomically and does not engage with the teaching as a whole. As is clear from the authorities cited above, the application must be considered overall, and if part of its teaching is general, so that would be clearly understood to apply generally, it is not necessarily added matter to take it in conjunction with another part of the general teaching.
300. In particular, although it is true that the passage at page 7 lines 26-29 refers to polysorbate 80 only, the reader would clearly understand that that was the example given at that stage, and not limiting, and would clearly appreciate that the patentee envisaged other surfactants, indeed a wide range of them, as equally applicable, given the teaching at page 17.
301. In my view the skilled reader would have all the above sections in mind and would clearly appreciate that surfactants generally, with many examples given, were useful to prevent silicone-induced aggregation in the context of the vaccines taught, with the formulation characteristics given.
302. I also think that this is a case where it is useful to ask what, if anything, is the additional information in the patent that is not in the application. Mr Tappin said in his opening that he perceived MSD’s case to be that the patent disclosed for the first time that it was not necessary for the surfactant used to be Tween 80. In due course, in his closing, Mr Hinchliffe agreed that this was the argument.
303. The fault in the argument is that while it is true that the Patent (by proposed amended claims 2 and 17) makes clear that it is not necessary for the surfactant used to be Tween 80, the application does not disclose (or even suggest) that it is necessary for the surfactant to be Tween 80. So there is no difference in teaching.
304. It seemed from the parties’ skeleton arguments in opening that there might be a deep divide over the applicable law. This turned out not to be the case, but it is still appropriate for me to set out the legal principles. I will do relatively briefly since I consider that the allegation fails on the facts in any event.
305. I have set out claims 1 and 16 above. Claim 1 is a product claim, and Wyeth accepts that if the physical features required are present then the claim lacks novelty, it being common ground as a result of the Order of Birss J to which I have referred that if the physical features are satisfied then the functional requirement for reducing silicone induced aggregation will be, too. That is why Wyeth did not resist the conclusion that Hausdorff 381 anticipates claim 1.
306. But Wyeth says that claim 16 is in a different category because it is a use claim. I have already said above that in my view the Order of Birss J did not have the effect of deeming the allegedly new use to be disclosed by the prior art, only of providing that the achievement of the effect in fact took place.
307. The fact that use claims are or may be different in this respect was established in MOBIL/Friction reducing additives (G02/88) [1990] EPOR 73. The decision of the Enlarged Board has caused much difficulty of understanding over the years, but Mr Hinchliffe did not submit that it ought not to be followed. The upshot is that a claim to the use of an old product for a new use is not anticipated by prior art which does not disclose the new use, even if performance of the prior art would inevitably have the technical result supporting the new use. Thus in MOBIL itself, prior art disclosing the use of the known products as a lubricant did not anticipate a patent to their use for inhibiting rust, even though persons who had put the prior art into effect would inevitably have been achieving rust reduction, without knowing it.
308. Analysis of MOBIL in the years since it was decided has focused, among other things, on the difficulties which it implies for infringement where people carry on doing what they were before the priority date of the contested patent but become fixed with knowledge that they are also achieving the new use, and how to decide whether the new use relied on is distinct from the use in the prior art.
309. The question of deciding whether a use is new or not is a potentially difficult one and the problems with understanding MOBIL are compounded by the fact that the uses in issue there (lubrication and rust inhibition) were so close to one another.
310. In his opening written skeleton, Mr Hinchliffe cited three TBA cases, as follows:
a) T279/93 for the proposition that the additional information in the patent in suit there did not teach the skilled person to do anything that they would not have done before without knowledge of the patent. If this is read too literally it would be inconsistent with the decision in MOBIL that second use claims of this kind are not anticipated by mere inevitable result without disclosure of the new use, but Mr Hinchliffe did not argue that;
b) T958/90 for the proposition that there is no novelty in disclosing that a known effect is present to a greater extent than previously understood; and
c) T892/94 for the proposition that a newly discovered technical effect does not provide novelty if it already underlay a known use of a known substance. In other words, a better understanding of a known use does not provide novelty.
311. He clarified in his oral opening that he did not contend that they qualified MOBIL in a relevant way, but rather were advanced to illustrate its application. In particular, he relied on the statement in T892/94 that providing new information about an old use did not meet the standard of providing a new use. He said that the relevant use in the present case was reducing aggregation and that the fact that it would (if not prevented) be caused by silicone was just information about that use. I return to this when I address the facts.
312. Leaving aside the MOBIL issues, the parties were agreed as to the overall standard for assessing novelty: clear and unambiguous disclosure.
313. As to the facts, MSD accepts that two claim features are not expressly disclosed in Hausdorff 381, namely:
a) whether the syringe is siliconized; and
b) the use to inhibit silicone induced aggregation.
314. As to a), Mr Hinchliffe stressed the CGK syringes were routinely siliconized, or otherwise they would not work. This would quite clearly be overwhelmingly strong evidence if the issue were one of obviousness, but there is a conceptual difference between anticipation and obviousness, not a difference of a degree. Strong obviousness does not lead seamlessly into anticipation; the tests are different. There is no disclosure of a siliconized container in Hausdorff 381 and so the novelty attack fails for this reason alone.
315. It fails for a further reason in relation to b). On this, MSD relied on combining a disclosure on page 15 of “a homogenous white suspension, ready for intramuscular administration” (which Prof Crommelin accepted taught that there was no material aggregation), with the disclosure of example 15 referring to aluminium phosphate. However, there is no disclosure that these passages are talking about the same thing. Again, a strong case could be made to combine these teachings if the issue were one of obviousness, but it is not. Paragraph 267 of MSD’s closing skeleton set out the nub of its reasoning, and to my mind it is very clearly an obviousness argument (even if a strong one) repackaged in the language of anticipation.
316. The fact that aluminium adjuvants were known as a matter of CGK to be capable, in some circumstances, of reducing aggregation does not mean that there is disclosure in Hausdorff 381 of using it in that way in Example 15 along with the physical features of the claim. There is no disclosure that were it not for the adjuvant, aggregation would occur.
317. This makes it strictly unnecessary to decide Mr Hinchliffe’s point that the requirement that the aggregation be silicone-induced cannot confer novelty. I will say that I agree with the point in principle: had there been disclosure of the use, in the context of a siliconized container, of an aluminium adjuvant to reduce aggregation, along with all the other features of claim 16, it would not be necessary to have a further disclosure that the aggregation was caused by the silicone: the goal and its achievement would be disclosed and the means by which the problem being addressed was caused would not be a new use.
318. De la Pena is a paper published in Spanish in 2004, from Wyeth’s workers in Madrid. It is about pneumococcal vaccination in Spain.
319. De la Pena refers (on page 50) to the non-conjugated 23v vaccine and to the 7v conjugated Prevenar. Both these are agreed to have been common general knowledge, as explained above.
320. At page 53 it refers to those two vaccines again, and then to three further conjugated vaccines which it says “have not been marketed and are in a very advanced stage of study”.
321. These are:
a) “The 9-serotype vaccine (it adds 1 and 5)”;
b) “- The 11-serotype vaccine (it adds 3 and 7F)”;
c) “- The 13-serotype vaccine (it adds 6A and 19A)”.
322. At page 53 right hand column there is the following further teaching about the 9-valent vaccine (referring to studies whose details are given on page 53 left hand column):
The study showed that using the PCV-9 vaccine prevents IPD, reduces pneumococcal resistance to antibiotics and decreases pneumonia in children. The remaining three studies (23, 24, 25) demonstrated the safety and immunogenicity of PCV-9, showing that:
- It is a safe vaccine and produced a good immune response to the 9 serotypes it contains.
- It is a well-tolerated vaccine
- There was a significant reduction in incidence of IPD caused by strains resistant to antibiotics.
- There was a decrease in nasopharyngeal carriers for vaccine pneumococci and an insignificant increase for nonvaccine pneumococci.
- There was a decrease in resistant pneumococcal carriers.
- Simultaneous administration of PCV-9 with routine vaccines in the immunisation programme is safe and immunogenic.
Therefore, we can say that PCV-9 is a safe, effective, and immunogenic vaccine that opens new possibilities in the field of conjugate vaccines.
323. And the following passage bridges pages 53-54:
The Future of Pneumococcal Vaccination
The 23-valent polysaccharide vaccine was the first step in fighting pneumococcal disease and the heptavalent conjugate vaccine has allowed us to drastically decrease the disease in younger children. With regard to future pneumococcal vaccination, several aspects must be borne in mind: serotypes and age and geographical distribution, combination with other vaccines, new routes of administration, and other strategies. The geographical variability of pneumococcal serotypes represents a problem when developing a vaccine with global coverage. There is almost a need to design a specific vaccine for each geographical area, having previously undertaken an epidemiological study of the most common serotypes, something only possible in developed countries. In addition, we know that the spectrum of serotypes broadens with age, which complicates the production of vaccines for age groups other than children, although they are the group at highest risk and in whom the current vaccine is most effective. In this regard, we are working on the incorporation of new serotypes to the PCV-7, and the nonavalent (incorporating serotypes 1 and 5), 11-v (additionally incorporating 3 and 7F) and 13-v (6A and 19A) vaccines are currently in different research phases, which could broaden the spectrum of ages and
countries, although coverage would remain very diverse. Furthermore, we are trying to incorporate the most antibiotic-resistant pneumococci.
324. There was no material dispute over the legal approach. I will consider obviousness using the 4 Pozzoli questions.
325. I have considered the skilled team and the common general knowledge above.
326. The steps from de la Pena to the claims of the Patent are as follows:
a) Select the 13v vaccine for progression;
b) Decide to use an aluminium salt adjuvant (filling in a gap, de la Pena is silent);
c) Decide to use a buffer (again, filling in a gap);
d) Decide to use a siliconized container (again, filling in a gap);
e) Add a surfactant to address silicone-induced aggregation; and
f) In order to make the decision to add a surfactant, the skilled team would have to observe that in the siliconized container, aggregation was occurring in the first place, since MSD’s case is, as I have already said, that a surfactant would not be added unless there was a purpose for it.
327. It is convenient to consider these steps from the point of view first primarily of the vaccinologist and then primarily of the formulator. I do this in order to try to bring structure to this judgment, and do not overlook that I am considering a team and that I have to see the picture as a whole, and not illegitimately slice the gap between the prior art and the invention into small pieces which may individually be, or seem, obvious.
328. As it happens, however, this overall task was eased because in closing Wyeth accepted (written closing paragraph 269) that if the skilled formulator was asked to progress the 13v vaccine of de la Pena, it would be obvious to use a siliconized container means, a pH buffered saline solution if required (with a pKa of about 3.5-7.5), and an aluminium adjuvant. These concessions were realistic to the point of inevitable, and it would have been better if they had been made much earlier.
329. One effect of this is that claim 1 is obvious if it was obvious to progress the 13v vaccine, but that is of little practical consequence.
330. I therefore consider first of whether it was obvious to progress the 13v vaccine, which is the vaccinology part of the picture.
331. Although de la Pena was of primary interest to a Spanish audience, it is not so limited. Similarly, it was probably of limited readership, but that does not mean it would not have been read with interest by the skilled vaccinologist.
332. The law requires that I consider it as if it were read with interest, and in this case that is not a fiction: it reports real practical work being done by a major company in the field with apparent success (to the extent of reaching “a very advanced phase of study”). The skilled vaccinologist would have had an active desire to progress the work reported. That would certainly, for infants, have involved using an aluminium adjuvant, as Wyeth concedes.
333. The real issue is whether the 13v vaccine specifically would have been an obvious choice to progress. The following factors are material:
a) The motivation to do so is spelled out on page 54: to broaden the spectrum of ages and countries covered, and deal with antibiotic-resistant pneumococci.
b) The skilled vaccinologist would know from the CGK that there was a strong case that the addition of 19A would give cross-protection and that there was a reasonable case that the addition of 6A could be useful from that perspective.
c) From de la Pena, the skilled vaccinologist would infer that Wyeth had been convinced of the cases for the addition of the 19A and 6A, sufficiently to put a lot of work into it.
334. The skilled vaccinologist would be aware that the technical issues would increase in difficulty with more valencies, but would have confidence that the 13v could be made to work from the report that Wyeth had successfully reached advanced studies.
335. However, the skilled vaccinologist would also have to consider the 9v and 11v as candidates for progression.
336. It was on the last of those points that Wyeth focused. It pointed out that in the period between the publication of de la Pena and the priority date of the Patent, Sanofi and GSK had both abandoned their 11v vaccines, with GSK deciding to take forward only a 10v vaccine. It also pointed out, based on the evidence of Prof Eskola, that nothing further would have been heard of the 13v vaccine in the 2004-2006 period.
337. I have dealt with the Sanofi/GSK matters in connection with the CGK, above. They do not support Wyeth’s position, or at least not much, because although those two companies had had difficulties the reader of de la Pena would know that Wyeth had overcome them so as to be able to do trials, and that the difficulties were therefore likely to be for reasons specific to those companies and not innate to the task.
338. If the skilled vaccinologist had decided to look into progression of the 13v vaccine (e.g. because they were spurred to do so having regard to their knowing of the Sanofi/GSK issue), it transpired from the cross-examination of Prof Eskola that they would have found a variety of references showing that it was being taking forward. So that point also fails on the facts. Mr Hinchliffe for MSD relied on these materials only as a shield to the point that the 13v vaccine might be thought to have been dropped, rather than actively asserting that they would have been found by someone considering what to do with de la Pena. Whether or not he needed to concede that, that is the basis on which I have proceeded.
339. Overall, it is my clear conclusion that it was obvious to progress the 13v vaccine from de la Pena.
340. It might also have been obvious to progress the other (9v and 11v) vaccines mentioned, and probably would have been, but it is perfectly possible for a piece of prior art to render multiple possibilities obvious. The skilled team would know that the 13v route could well be more challenging technically, and I bear that in mind, but I consider the prospects of success weighed against the likelihood of achieving better protection would be more than sufficient to justify the work. There would be a high degree of confidence that provided the technical issues inherent in formulating the 13v vaccine could be addressed (where Wyeth’s progress was encouraging, as I have said), the countervailing benefits of better protection against more serotypes, better geographic coverage and better utility against antibiotic resistant strains, compared with the 7v, 9v or 11v vaccines, would be achieved.
341. I then have to consider the formulator’s task if asked to progress the 13v vaccine from de la Pena, to identify what it would be obvious to do without invention. As I have explained above, most of the formulation requirements of the claims were accepted to be routine if it was decided to pursue the 13v vaccine from de la Pena. The analysis as to the disputed features follows quite readily from the findings I have made in relation to CGK:
a) Aggregation would in fact take place.
b) Aggregation would be observed by routine work, either by regular testing or when the vaccine was put into commerce and, for example, agitated in shipping. It would in fact be caused by the silicone in the final containers (the use of which was accepted to be obvious).
c) The notional skilled formulator would readily appreciate that the cause of the aggregation was the silicone.
d) One obvious way to address the aggregation would be a surfactant, and one obvious surfactant would be polysorbate (20 or 80).
e) The expectation of success would be high because it would be using a known thing for one of its known purposes, in a known context.
342. It is not my finding that a surfactant would be seen as the only way to go. The skilled team and the formulator in particular would consider other changes such as trying a different buffer, or tweaking the concentrations of the excipients.
343. The Wyeth work supports this: they tried a number of different things. This is a factor to be taken into account and I do so, but it does not change my conclusion. I think there were several obvious routes.
344. Indeed, overall the Wyeth story, which was presented and gone into at a much greater level of detail than that at which arguments about obviousness usually proceed, left me with the clear impression that although the workers were initially somewhat thrown by the particular way in which the aggregation problem had been first missed and then found, thereafter they proceeded without drama and by systematic work to the decision first to try and then to use, a surfactant. The fact that the story was presented in such detail does not demonstrate invention or even unusual complexity. There were many small steps in what they did but that is simply because in this field the work is careful and systematic.
345. Standing back, as I consider I should, I think that the claims of the Patent are about taking forward a very attractive proposal (the 13v vaccine in de la Pena) by routine means, including solving a modest CGK problem (aggregation caused by silicone) in a way which was CGK (a surfactant).
346. The claims are all therefore obvious over de la Pena.
347. Chiron is a patent application filed in 2002 and published in February 2003.
348. Chiron was a well known company in the field of vaccines and the reader would readily appreciate this.
349. The attack based on Chiron must stand or fall on its own, independently of de la Pena. They cannot legitimately be combined. Nonetheless, I can conveniently explain my reasoning by referring back to what I have said about de la Pena.
350. Chiron is really a document about vaccine formulation. Its idea is to use histidine as a buffer in combination with an aluminium salt.
351. Because it is really a formulation teaching, Chiron does not say anything specific about the immunological characteristics of the vaccine(s) for which it might be used. It merely contains a general disclosure about possible vaccine antigens in a long list over pages 2 and 3. The list does include Streptococcus pneumoniae. Pneumococcal disease is in a preferred list of applications on page 6 among a number of others, but this is a weak pointer.
352. Chiron provides no teaching about which serotypes to use, if the skilled person were to focus on pneumococcal vaccines. It contains no pointer to the 13v combination of the claims of the Patent.
353. As a result, MSD had to make its case on the vaccinology side from CGK alone. This involved (as I understood it) the following proposed steps:
a) Choosing to take Chiron forward for pneumococcal disease.
b) Getting to the 11v vaccine (lacking 6A and 19A) as a starting point; and
c) Adding 6A and 19A.
354. This is a quite different situation from that which arises with de la Pena. That document had a clear focus on pneumococcal disease, a specific teaching of the 13v combination, a rationale for undertaking it, motivation, and a statement that real work had been done and real progress had been made by a leading player. Chiron lacks any of these, and on top of that it does not even provide the 11v vaccine as platform from which to start: the skilled team would have the uncertainty about whether that could be achieved without trouble.
355. Mr Hinchliffe submitted that coming up with the 13v combination just meant using each of the serotype antigens for its known purpose, and that each would be expected to provide some degree of protection. That is true, but it is not an adequate argument for obviousness. If it were accepted, any and every combination would be obvious. Yet real work and thought clearly went into making choices of combinations of valencies: it simply was not possible to choose any combination that one wanted, otherwise (as both sides pointed out) the answer would be to include every single one. Protection, including cross protection, had to be balanced against the practical limit on how many serotype antigens could be included.
356. As to the formulation side of the matter, Chiron does contain a teaching, to which I have referred above, at page 6 lines 14 to 15, that “The composition may comprise a detergent (e.g. a Tween such as Tween 80) in order to minimise adsorption of antigens to containers”. In view of my findings on common general knowledge this supports but would not be necessary to MSD’s case. Otherwise, the formulation attack is the same as over de la Pena. Had it been obvious to progress the 13v pneumococcal option from Chiron, the inclusion of a surfactant would have been obvious for all the same reasons.
357. Since I have held the Patent obvious over de la Pena this does not arise, but I will address it briefly.
358. MSD says that the Patent is insufficient because if the 13v combination was not obvious, there is nothing in the Patent’s teaching to make it plausible that that combination would be efficacious (or safe). The attack focuses very much on the vaccinology side of the case. It was also put as a form of obviousness based on lack of technical contribution, but that is just a different legal description of the same attack and does not add anything.
359. MSD cited the following passages from the judgment of Arnold J as he then was in Generics v. Yeda [2018] RPC 2:
191. The claimants contend that, if the claimed inventions were not obvious over the prior art, then there is nothing in the Patent which makes it plausible that 40 mg TIW would be efficacious compared to placebo or as efficacious as 20 mg QD or more tolerable, because the Patent contains neither any clinical data nor any theoretical justification for such a supposition beyond what was already known to the skilled person. Accordingly, the claimants contend that the claimed inventions make no technical contribution to the art and are therefore do not involve an inventive step in accordance with the principles summarised by Floyd L.J. in Generics (UK) Ltd v Yeda Research and Development Co Ltd [2013] EWCA Civ 925, [2014] R.P.C. 4 at [49], alternatively are insufficient in accordance with the principles summarised by Kitchin L.J. in Idenix Pharmaceuticals Inc v Gilead Sciences Inc [2016] EWCA Civ 1089 at [137]-[138]. The principles applied by the English courts are based on those developed by the Boards of Appeal of the EPO, which are well illustrated by two recent contrasting decisions of the same Board relating to dasatinib in Cases T 488/16 (1 February 2017) and T 950/13 (3 February 2017).
…
193. Counsel for the Claimants accepted that the legal tests to be applied were different. He submitted that this did not matter on the facts of the present case because all that the Defendants could rely upon to establish that the claimed inventions were not speculative was the skilled person's common general knowledge outlined above. If 40 mg TIW was not obvious to try with a fair expectation of success in the light of the skilled person's common general knowledge, then the claim that 40 mg TIW would be efficacious compared to placebo was speculative and the claim that it would be as efficacious and more tolerable than 20 mg QD was even more speculative.
…
195. In my judgment, if the claimed inventions were not obvious, then the claim that 40 mg TIW would be efficacious compared to placebo was credible, but the claim that it would be as efficacious and more tolerable than 20 mg QD was speculative. I therefore conclude that claim 3 would be invalid for lack of an inventive step and insufficiency, but not claim 1 as I have construed it.
360. MSD also relied on a similar passage, again from Arnold J, in Allergan v. Aspire [2019] EWHC 1085 (Pat) at [125].
361. Mr Hinchliffe accepted the statement in Generics v. Yeda that the standards are not the same for insufficiency and obviousness, and he also accepted (indeed he cited) the proposition (from Warner-Lambert v. Generics [2018] UKSC 56 at [36]-[37]) that the standard for plausibility is a low one, and plausibility may in some situations be demonstrated by a priori reasoning without experimentation.
362. These cases establish that squeezes of this kind can in principle work, but it depends on the facts. They work better the less that the patent under attack provides that is new over the prior art, and best when it provides nothing.
363. In the present case it simply was not supported by evidence that the 13v combination of the claims was not plausible. MSD’s positive case was that it was. Wyeth’s position was that it was plausible once thought of, and that is part of the reason why I have found the Patent obvious over de la Pena (which identifies it specifically) and not over Chiron (which does not identify it at all). It was not inconsistent for Wyeth to say that without it being pointed to, and a case for it made, it would not be obvious to think of it and then pursue it.
364. So far as the Patent’s teaching is concerned, it is true that there is no experimental proof of the protective effect of the 13v combination, but the law does not require it. The combination is clearly identified in the specification and although the concrete work disclosed is about formulating the combination and protecting it from aggregation, that is not immaterial to the achievement of the claims, because as I have explained above, part of the challenge in this field is to balance the multiple serotype antigens that would be desirable with the practical challenges of making the vaccine.
365. I therefore reject the insufficiency attack which was in any event only a squeeze.
366. My conclusions are:
a) The allegation of infringement of claims 1 and 16 and all dependent claims fails.
b) I decline to decide the subsidiary allegation of infringement by equivalence since it is rendered irrelevant by my other conclusions.
c) Claims 2 and 17 do not add matter.
d) Claim 1 is anticipated by Hausdorff 381 (a conclusion against which Wyeth mounted no defence), but claim 16 is novel over it.
e) All the claims are obvious over de la Pena.
f) The allegation of obviousness over Chiron fails.
g) The insufficiency attack fails.
367. I will hear Counsel as to the form of order, if it cannot be agreed.
