B e f o r e :
HIS HONOUR JUDGE HACON
(Sitting as a High Court Judge)
____________________
Between:
| |
(1) SANDOZ AG
(2) SANDOZ LIMITED
(3) ACCORD HEALTHCARE LIMITED
|
Claimants/Part 20 Defendants in HP-2022-000029
|
| |
(4) TEVA PHARMACEUTICAL INDUSTRIES LIMITED
|
Claimant/Part 20 Defendant in HP-2022-000032
|
| |
(5) CIPLA LIMITED
|
Claimant/Part 20 Defendant in HP-2022-000034
|
| |
(6) AMAROX LIMITED
(7) HETERO LABS LIMITED
|
Claimants/Part20 Defendants in HP-2023-000005
|
| |
(8) GENERICS (UK) LIMITED
(9) VIATRIS (UK) HEALTHCARE LIMITED
|
Claimants/Part 20 Defendants in HP-2023-000006
|
| |
(10) STADA ARZNEIMITTEL AG
|
Claimant/Part 20 Defendant in HP-2023-000017
|
| |
- and -
|
|
| |
BAYER INTELLECTUAL PROPERTY GmbH
|
Defendant/Part 20 Claimant
|
| |
- and -
|
|
| |
(1) TEVA (UK) LIMITED
(2) CIPLA (EU) LIMITED
(3) THORNTON & ROSS LIMITED
(4) GENUS PHARMACEUTICALS LIMITED
|
Third Parties
|
____________________
Adrian Speck KC, Henry Ward, Adam Gamsa and Michael Conway (instructed by Pinsent Masons LLP, Bristows LLP, Penningtons Manches Cooper LLP, HGF Law LLP and Taylor Wessing LLP) for the Claimants/Part 20 Defendants and Third Parties
Andrew Waugh KC and Alice Hart (instructed by Allen & Overy LLP) for the Defendant/Part20 Claimant
Hearing dates: 9, 12-16 and 21-22 February 2024
____________________
HTML VERSION OF JUDGMENT APPROVED
____________________
Crown Copyright ©
Judge Hacon :
Introduction
- Rivaroxaban is the generic name of a pharmaceutical� used for the prevention or treatment of thromboembolic disorders. It was developed by the Bayer group of which the defendant forms part. I will refer to the defendant and other members of the group individually and collectively as "Bayer".
- The active ingredient of rivaroxaban is protected by product claims of a patent owned by Bayer. Consequently Bayer has had a monopoly of the rivaroxaban market, selling the product under its brand name Xarelto. That protection was extended by an SPC but expired on 1 April 2024. This provides an opportunity for those who wish to market generic rivaroxaban, not least the claimants and third parties in these proceedings, save that Bayer also owns European Patent (UK) No. 1 845 961 ("the Patent").
- The Patent has claims in Swiss form covering the use of the active ingredient for the manufacture of a medicament for the treatment of a thromboembolic disorder. The claims are limited, so far as is relevant, to (a) the use of a rapid release tablet and (b) administration no more than once daily for at least five days. There is no limitation as to the dose. The priority date of the Patent is 31 January 2005.
- The claimants allege that the Patent is invalid and that it constitutes an attempt by Bayer to evergreen its lucrative monopoly in the manufacture and marketing of rivaroxaban, in other words to extend the monopoly through unjustified means.
- Bayer has counterclaimed, alleging a threat on the part of the claimants and the third parties to infringe the Patent by the marketing of rivaroxaban products.
- All six actions joined into the present proceedings were brought by one or more of the claimants, seeking revocation of the Patent. In every action Bayer counterclaimed alleging a threat to infringe the Patent, in some instances joining one or more third parties as alleged potential infringers. It is not necessary for me to distinguish between the claimants and third parties which I will collectively refer to as "the claimants". The threat to infringe was in all instances conceded for the purposes of this trial. This action is solely about the validity of the Patent.
- Adrian Speck KC, Henry Ward, Adam Gamsa and Michael Conway appeared for the claimants, Andrew Waugh KC and Alice Hart for Bayer.
Technical background
- The following matters all formed part of the skilled person's common general knowledge ("CGK") at the priority date.
Haemostasis and thromboembolism
- Haemostasis is a process which gives rise to the formation of blood clots when an individual suffers injury and there is a need to close up damage to blood vessels. Haemostasis is a necessary local response to injury but vascular blood should otherwise flow freely. In healthy humans there is therefore a homeostatic balance, meaning a self-regulating process which adjusts to prevailing conditions to best suit the individual's survival, between procoagulant and anticoagulant mechanisms.
- Thrombosis is a condition in which a clot, or thrombus, causes obstruction of the blood flow in part of the vascular system away from a site of injury. Thrombosis has long been treated by the administration of anticoagulants.
The coagulation cascade
- Blood has a liquid component, plasma, and three solid components: red blood cells, white blood cells and platelets. The primary purpose of red blood cells is to carry oxygen but they have a secondary function in that they become passively trapped in thrombi, forming the bulk of a thrombus. The primary purpose of white blood cells is to combat infection. Some classes of one type of white blood cell, leukocytes, express on their surface a protein called "tissue factor" during haemostasis which activates blood coagulation. Platelets are very small cell fragments which, when activated, aggregate and adhere to nearby surfaces, forming part of a thrombus.
- The "coagulation cascade" is the name given to a series of reactions culminating in the formation of an insoluble clot. The enzymes which catalyse the reactions are known as "factors", identified by Roman numerals and given the suffix "a" when the factor is in its active form. The following provides an overview of the coagulation cascade:
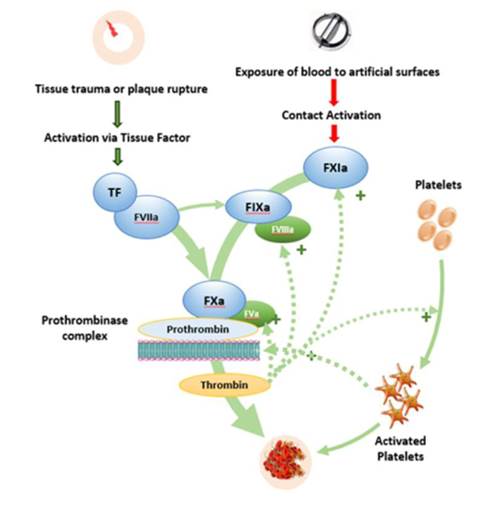
- The upper part of the diagram shows two pathways, each contributing and leading to the activation of Factor X to Factor Xa ("FXa"). The one on the left is known as the extrinsic pathway, the one on the right is the intrinsic pathway. FXa in association with Factor Va converts prothrombin (Factor II) to thrombin (Factor IIa). Thrombin feeds back into the cascade to activate other factors and platelets, an effect shown by the dotted lines. Thrombin also converts fibrinogen to fibrin. Fibrin is an insoluble polymer in the form of threads which stabilize the structure of a clot.
Thromboembolic disorders
- The primary causes of thrombosis are the improper functioning of the coagulation cascade and/or excessive platelet activation, commonly because the anticoagulation system is overwhelmed by thrombotic stimuli. A thrombus may break loose and be carried elsewhere in the circulatory system. In that form it is known as an "embolus", the condition is "embolism". "Thromboembolism" describes the combined conditions of thrombosis and embolism.
- Patients undergoing major surgery are at particularly high risk of venous thromboembolism ("VTE"). One form is deep vein thrombosis ("DVT"), the most serious complication of which is pulmonary embolism ("PE") in the lung. PE remains a significant cause of death in both surgical and medical patients.
- The human body has its own mechanisms to modulate clot formation. There are three, each inactivating factors in the coagulation cascade and in one case also inactivating thrombin. Where urgent anticoagulation in a patient is needed, drugs are used to prevent the formation of thrombi and/or to degrade them.
The therapeutic window
- An important feature of anticoagulant drugs is that if they are not sufficiently effective, clots will remain and new clots may form. If they are too effective, clots required at the site of injury will not form, particularly after surgery, and the patient will suffer excessive bleeding. Both possibilities are dangerous for the patient and either may be fatal. There is thus a "therapeutic window" in which the drug is available enough to have a sufficiently anticoagulant effect, but not so available and effective such that the patient suffers from unwanted bleeding.
- The wider the therapeutic window afforded by a drug, the more attractive it is for clinical use. But it is never possible to know in advance where the therapeutic window lies and how wide the window will turn out to be.
Prior art anticoagulant drugs
- The following anticoagulant drugs were in clinical use at the priority date of the Patent, 31 January 2005:
(1) Warfarin is an orally administered vitamin K antagonist. It inhibits a liver enzyme that blocks the recycling of vitamin K, which has the effect of reducing the production of Factors II, VII, IX and X in the liver. First marketed as a rat poison, it was approved for use in humans as an anticoagulant in the 1950s. It is relatively slow-acting and its effect varies between patients, so close monitoring and dose adjustment is necessary. However it remains widely prescribed in the UK and elsewhere.
(2) Heparin indirectly inactivates thrombin and FXa. There are two forms, unfractionated heparin, usually referred to in the evidence as just "heparin", and low molecular weight heparin (LMWH). Both are administered by intravenous or subcutaneous injection. Heparin, like warfarin, exhibits variability in its effect and so requires patient monitoring and dose adjustment. LMWH is more predictable in its effect so no patient monitoring is required. Enoxaparin is a commonly used LMWH.
(3) Fondaparinux also indirectly inhibits FXa. It is administered subcutaneously once daily. By January 2005 it had been evaluated extensively for the prevention of VTE in patients undergoing orthopaedic surgery, i.e. surgery concerned with conditions involving the musculoskeletal system. It was found to be more effective than enoxaparin in preventing post-operative thrombosis.
- Three other anticoagulants had been licensed for treatment by January 2005, namely hirudin, bivalirudin and argatroban. They featured less in the evidence.
- Included in CGK was an awareness that several new anticoagulants were in development:
(1) Ximelagatran is a direct thrombin inhibitor, orally administered twice daily. Promising results from clinical trials encouraged the expectation that it would provide a safe orally administered anticoagulant. It has not been marketed due to the emergence of issues concerning liver toxicity but these were not known in January 2005.
(2) Idraparinux is an indirect FXa inhibitor administered subcutaneously once weekly. Clinical trials suggested that a relatively high dose could cause fatal bleeding, but trials were being pursued with a lower dose.
(3) Razaxaban is a direct FXa inhibitor, orally administered twice a day. Trials of higher doses had been discontinued because of excessive bleeding.
(4) DX9065a is a direct FXa inhibitor administered by continuous intravenous infusion. It was undergoing a small trial in January 2005.
Clinical trials
- Drugs undergoing development are subjected to pre-clinical tests which, if successful, will lead to clinical trials. All are mandatory and governed by rules issued by regulatory bodies. Pre-clinical trials include tests as to safety, toxicity and what are sometimes called "proof-of-concept" studies which give an early indication of whether the drug will have the intended effect.
- Tests will be in vitro or ex vivo and may also involve experiments on animals.
- Clinical trials are carried out in three phases, I to III. Phase I clinical trials are conducted on a small number of healthy volunteers. At the priority date they were almost invariably young healthy males, although one of the clinical experts said that more recently there has been a move to greater diversity of risk and healthy female volunteers have been recruited. The primary goal is to obtain data on safety, toxicity and what are called pharmacokinetics (PK) and pharmacodynamics (PD). Broadly, the former measure the effect of the body's system on the drug over time, the latter the effect over time of the drug on the body's system.
- The key PK measurement is the half-life of the drug. Following administration, the drug may be metabolised to an inactive form and will be excreted. Its concentration in blood plasma therefore declines. The rate of decline varies between drugs. As the term implies, the half-life of a drug is the time taken for its content in the plasma to decline by half. The PK profile is a graphical representation of the plasma concentration against time from initial administration until it reaches zero. The cumulative exposure of the individual to the drug is thus shown by the area under the curve or AUC.
- PD data measure the effect over time that the drug has on the patient, using "bio-markers" which are measurable indicia associated with the intended effect of the drug. Below there are examples of these for anticoagulants.
- There may or may not be a correlation between the half-life of a drug and any one or more PD measurements. Thus, the level recorded from the bio-marker assay may not decline by half at the same time that the drug concentration has reached its half-life. To a greater or lesser extent PD data may follow a curve different to the PK curve.
- The first phase I study is usually carried out with the administration of successively ascending single doses. Single ascending dose studies were referred to in the evidence as "SAD studies". Typically these are followed by administering multiple ascending doses, "MAD studies".
- If the phase I trial or trials are successful, the pharmaceutical research team will move on to phase II. The cohort of individuals recruited is larger and unlike those in phase I they will all have the condition to be treated by the drug being tested. In the case of an anticoagulant, they will typically be patients who have had surgery to replace a knee or hip and who are vulnerable to VTE. The dosage and the frequency of administration will be informed by the results of the phase I trial.
- If the phase II trial reveals one or more regimens of dosage and frequency of administration which gives satisfactory results, these will be used for phase III studies for which a still larger cohort of patients with the relevant condition are treated.
Coagulation assays used for PD studies
- Several assays have been developed to measure the effect of anticoagulants. These are used in pre-clinical studies as well as for obtaining PD data in phase I clinical studies. They are, or at least include:
(1) Prothrombin time or "PT". This measures the time taken to generate fibrin via the extrinsic pathway following activation of Factor VII by tissue factor. It is the test used to monitor the dosage of warfarin. PT is reported as an international normalized ratio or INR.
(2) Activated partial thromboplastin time or APTT. This measures the time taken to generate fibrin by the intrinsic pathway. Activators of factors in that pathway are used to cause the creation of a clot, subject to addition of the coagulant under test which will prolong that time. The time taken is the APTT. APTT was generally used to monitor the activity of heparin.
(3) The Hep Test. An excess of FXa is added to the test plasma containing the anticoagulant and to control plasma. After incubation the mixtures are recalcified and the clotting time recorded. It is used to measure levels of heparin and LMWHs and could be adapted to measure direct FXa inhibitors. At the priority date it was not routinely used for patient monitoring but may have been used for research.
(4) FXa assay. A plasma sample is drawn from a subject who has taken an anticoagulant which operates as a FXa inhibitor. The residual FXa left uninhibited is measured using a chromogenic substrate specific for FXa. This was only available in specialised laboratories used for research (such as those run by Bayer), as opposed to labs used for clinical patient monitoring.
- Three other assays were mentioned in the evidence: endogenous thrombin potential (ETP), prothrombinase-induced clotting time assay (PICT) and platelet-induced thrombin generation time (PITT).
- All the foregoing assays measure changes in a biomarker associated with anticoagulation and the effect over time of the drug being tested on that biomarker. The tests are typically carried out ex vivo, which means that successive samples of blood are taken from an individual to whom the drug has been administered and changes in the biomarker are monitored in samples taken over an appropriate period.
- At the priority date none of these had been established as guaranteed predictors of efficacy or safety for direct FXa inhibitors. Nonetheless they were considered useful enough indicators to use in phase I trials of a potential antithrombotic drug. For a drug known to act by inhibiting FXa, assays that measure FXa level were the most useful.
- Measuring anticoagulation effect in this way is not a direct means of measuring antithrombotic effect, neither in the sense of prophylaxis preventing the formation of thrombi, nor treatment removing thrombi. That can only be done by testing the drug in a patient suffering from or vulnerable to thrombosis, which happens in a phase II or phase III trial. Typically this is done using a venogram scan. The scan involves the injection of a contrast dye into the veins under investigation and then examination of the veins by X-ray.
The Patent
- The title of the Patent is "Treatment of thromboembolic disorders with rivaroxaban". The specification refers to the development of drugs which inhibit FXa for the treatment and prophylaxis of thromboembolic disorders. It continues:
"[0009] In general, oral application is the preferable route of administration of a drug, and a less frequent dose regimen is desirable. In particular, once daily oral application is preferred due to favourable convenience for the patient and for compliance reasons. However, this goal is sometimes difficult to achieve depending on the specific behaviour and properties of the drug substance, especially its plasma concentration half life.
[0012] Surprisingly, it has now been found in patients at frequent medication that once daily oral administration of a direct FXa inhibitor with a plasma concentration half life time of 10 hours or less demonstrated efficacy when compared to standard therapy and at the same time was as effective as after twice daily (bid) administration."
- The Patent contains a single experimental example, being a clinical study of rivaroxaban, identifiable as such from paragraph [0014] which sets out the chemical structure of the active ingredient. The purpose, set out from [0035], was to assess the safety, tolerability, and efficacy of rivaroxaban at different oral doses, administered twice or once daily, compared with 40 mg of subcutaneously administered enoxaparin in the prevention of venous thromboembolism. 642 patients were enrolled, men over 18 years of age and postmenopausal women, all undergoing elective primary hip replacement. It was common ground that this would he recognised as a phase II clinical trial.
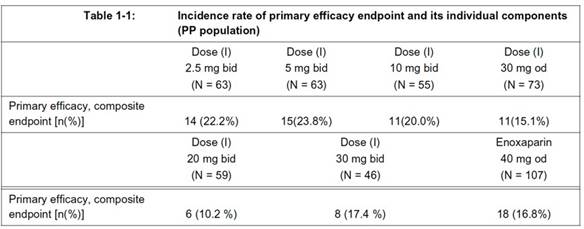
- The Patent states that patients either received 40 mg of the comparator, enoxaparin, once daily, or received rivaroxaban according to one of the following regimens: 2.5 mg twice daily, 5 mg twice daily, 10 mg twice daily, 20 mg twice daily, 30 mg twice daily or 30 mg once daily. The duration of the trial was 7-9 days.
- Two tables are presented. The first sets out what are called efficacy results, the percentage reduction in VTE incidence rates. This is the table, where (I) means rivaroxaban, bid means twice a day, od means once a day and N provides the number of patients for each regimen:
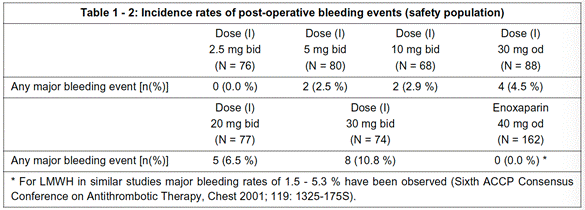
- The second table provides safety results in the form of percentage incidence rates of post-operative bleeding:
- Beneath the second table there is a summary:
"Summary: The above data clearly demonstrate the efficacy of od administration of (I), namely fewer occurrence of composite endpoint events, i.e. fewer cases of DVT, PE or death compared to untreated conditions, and in the range of standard therapy. Furthermore, the od administration is surprisingly perfect in line with bid administration."
- These are claims 1 and 2:
"1. The use of a rapid-release tablet of [rivaroxaban] for the manufacture of a medicament for the treatment of a thromboembolic disorder administered no more than once daily for at least five consecutive days, wherein said compound has a plasma concentration half life of 10 hours or less when orally administered to a human patient.
2. The use as claimed in Claim 1, wherein the thromboembolic disorder is ST Segment Elevation Myocardial Infarction (STEMI), Non ST Segment Elevation Myocardial Infarction (NSTEMI), unstable angina, reocclusion after angioplasty or aortocoronary bypass, pulmonary embolisms, deep vein thromboses or stroke."
The skilled team
- It was common ground that the skilled team primarily consisted of a pharmacologist and a clinician interested in the treatment of thromboembolic disorders. It was agreed that the two members of the team would work closely together when forming a composite view of the prior art.
- It was also common ground that the skilled team would call upon the expertise of an information specialist if needed. The specialist in question would be familiar with the process of finding from published sources information about research conducted by pharmaceutical companies.
The witnesses
- Each side filed evidence from an expert clinician and an expert pharmacologist.
- The claimants' expert clinician was Professor Jack Hirsh. He is Professor Emeritus in the Department of Medicine at McMaster University in Hamilton, Ontario. He has had a long and distinguished career which has included pioneering the use of warfarin in a low intensity treatment and promoting an international standardisation of units for measuring such treatment. He has particular expertise in LMWHs. His work led to the regulatory approval of enoxaparin, an LMWH.
- Professor Martin Wilkins provided the claimants' expert evidence on pharmacology. He is Professor of Clinical Pharmacology at Imperial College, Director of the British Heart Foundation Imperial College Centre of Research Excellence and Director of the National Institute for Health and Care Research at Hammersmith Hospital. Professor Wilkins has practised in clinical pharmacology for over 40 years in Birmingham and London during the course of which he has participated in many clinical trials.
- Professor Mark Crowther was Bayer's expert clinician. Like Professor Hirsh he is an academic based at McMaster University, being Professor and Chair of the Department of Medicine and Leo Pharma Chair in Thromboembolism Research. His work includes research and clinical practice relating to the management of thromboembolism in patients. He is a fellow of the Canadian Academy of Health Sciences.
- Bayer's expert on pharmacology was Professor Bernd Meibohm. He is Distinguished Professor of Pharmaceutical Sciences, Chair of the Department of Pharmaceutical Sciences, Harriett S. Van Fleet Endowed Professor in Pharmaceutics and the Associate Dean for Research and Graduate Programs at the College of Pharmacy of the University of Tennessee Health Science Center in Memphis, Tennessee. Professor Meibohm conducts research into the optimization of dosing regimens, adapted to the needs of the individual patient.
- None of the experts was an approximation of his corresponding person skilled in the art, all were too qualified. I do not believe that this prevented any of them from doing his best to adopt the mindset of the relevant skilled person at the priority date.
- It is not unusual for expert witnesses to know more than the skilled person when giving their evidence; the question is whether they are able to exclude such knowledge from their reasoning where this is needed. Where this arises and is relevant to the issues in the trial, it is important that the expert identifies his or her prior knowledge. Both sides criticised experts on the opposing side for not having done this.
- In a sustained part of his cross-examination it was put to Professor Hirsh that when he was giving his evidence he must have known that the case concerned rivaroxaban and that his knowledge of the drug's characteristics had influenced that evidence. Professor Hirsh was very clear that he did not know and I accept that evidence. It emerged that Professor Crowther had referred to rivaroxaban's known half-life when giving evidence in Canada in 2016 and so knew it, or at least had known it, when preparing his evidence in these proceedings. Professor Crowther was clear that by the time of drafting his evidence here he had forgotten the figure and I accept this too. Finally, Bayer submitted that Professor Wilkins should have said in his report that he knew that rivaroxaban was administered once daily. In cross-examination he agreed that he could well have known at some time, but by the time of preparing his evidence he would have had to look that up. I am satisfied that there was nothing of substance in the criticism.
- All four experts gave careful consideration to the questions put to them and for much the greater part gave clear and direct answers. They differed on important issues in the case, each in line with the party by which they had been instructed, but I do not doubt that each was doing his best to provide technical assistance and appropriate opinions to the court according to their instructions.
- There was evidence of fact from a former Vice-President and Head of Therapeutic Area of Cardiovascular and Coagulation at Bayer, Dr Frank Misselwitz. Like the experts on researching information, his evidence went to a narrow point, better discussed in context.
Hindsight and consultation between the experts
- Bayer had three criticisms of the way in which Professors Hirsh and Wilkins had prepared and presented their evidence.
- The first was that they had been insufficiently candid in their reports about what they knew about BAY 59-7939 when considering the prior art. I have discussed this above. There was nothing of importance which had not been said in their reports.
- The second criticism was that both were first asked to consider Harder, a cited item of prior art, by itself and only later were given the Kubitza posters, taken to be read together with Harder for the reason discussed below. I think there is nothing in this either. There was a point between the parties as to whether Harder should be treated as an item of prior art without reference to the Kubitza posters. It was not unreasonable for the witnesses first to consider Harder in isolation. They then gave their written evidence about the combined disclosure of Harder plus the Kubitza posters.
- There is a little more substance in the third criticism. It was that whereas in real life a clinician and pharmacologist working in a team on a project to find an appropriate dosing regimen for a drug would work together from the start of the project, Professors Hirsh and Wilkins did not consult each other. Instead, each saw a draft of the other's report shortly before it was filed. To some extent the same criticism could be levelled to some degree at how Professors Crowther and Meibohm were instructed to prepare their evidence, although they looked at a draft of each other's evidence earlier in the process, not just before filing.
- It has become established that where real life experts would collaborate in the pursuit of a project, corresponding expert witnesses should consult one another in a broadly similar way, see Alcon Eye Care UK Ltd v AMO Development LLC [2022] EWHC 955 (Pat) at [233]-[235], Teva Pharmaceutical Industries Ltd v Novartis AG [2022] EWHC 2847 (Pat) at [38]-41] and Teva Pharmaceutical Industries Ltd v Grünenthal GmbH [2023] EWHC 1836 (Pat) at [51]-[52]. That should have happened in the present case.
- The main consequence of it not happening, as submitted by Bayer, was that Professor Wilkins gave ill-informed evidence. First, he did not have a full appreciation of the consequences of underdosing and overdosing in a phase II trial and so could not provide reliable evidence as to the skilled person's reasonable expectation of the safety and efficacy of any dosing regimen. Secondly, he did not know that there was no established correlation between FXa inhibition assays and therapeutic safety and efficacy. Thirdly, he did not know the clinical significance of animal experiments referred to in Perzborn, another cited item of prior art.
- As to the first, Professor Wilkins said that he was aware of the consequences of underdosing and overdosing but agreed that he did not know what degree of caution would be appropriate, this was a matter for the clinician. To the extent that Professor Wilkins made an estimation about this when drafting his evidence, it was not shown that he was wrong.
- Regarding the second point, it seems to me that Professor Wilkins was entitled to assume that the PD assays in the prior art would be treated as providing at least some indication of the therapeutic safety and efficacy of BAY 59-7939. That was common ground. He interpreted the results from a biological perspective as opposed to a clinical one, but that did not make his conclusions meaningless.
- As to the third, he said in cross-examination that he gave an interpretation of the biological significance of the animal tests of Perzborn, not the clinical significance. He had expressly stated in his report that this was the limit of his perspective on the prior art and was what would be taken from it by a skilled pharmacologist. If relevant parts of his evidence had been shown to be inconsistent with a view taken by Professor Hirsh, such evidence may have been of limited value. No such inconsistencies were pointed out.
- In my opinion the lack of consultation between Professors Hirsh and Wilkins, and to a lesser degree Professors Crowther and Meibohm, was more a problem of procedural form than evidential substance. On both sides each expert did consider his colleague's draft evidence and had the chance to alter what he had said in his own report. It was not shown that doing this belatedly had had a significant effect on the evidence given. On each side the opinions of the clinician and pharmacologist were in agreement on the key issues.
The cited prior art
- This is the cited prior art as pleaded in the Re-Re-Amended Grounds of Invalidity:
(1) The poster entitled "Effects of Bay 59-7939, an Oral, Direct FXa Inhibitor, on Thrombin Generation in Healthy Volunteers", by Sebastian Harder et al., ("Harder") which was made available to the public at the 45th Annual Meeting of the American Society of Hematology in San Diego, USA, held between 6-9 December 2003.
(2) The paper entitled "In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939 an oral, direct FXa inhibitor", authored by Elizabeth Perzborn et al., ("Perzborn") which was published online on 26 January 2005.
As cited, this was to be read together with
(a) "Single dose escalation study investigating the pharmacodynamics, safety, and pharmacokinetics of BAY 59-7939 an oral, direct FXa inhibitor in healthy male subjects." Blood 2003; 102: Abstract 3010, Kubitza D., et al., ("Blood 3010") and
(b) "Multiple dose escalation study investigating the pharmacodynamics, safety, and pharmacokinetics of BAY 59-7939 an oral, direct FXa inhibitor in healthy male subjects." Blood 2003; 102: Abstract 3004 ("Blood 3004").
Further or alternatively, Perzborn, Blood 3010 and Blood 3004 would been seen and read in combination by the skilled person with
(c) "Effects of BAY 59-7939, an oral, direct FXa inhibitor, on thrombin generation in healthy volunteers". Blood 2003; 102: Abstract 3003 ("Blood 3003"),
which is on the same page as Blood 3004.
I will call Bloods 3010, 3004 and 3003 collectively "the Blood Abstracts".
- All cited items were created and published by researchers working with or for Bayer on the early stages of Bayer's rivaroxaban project.
- The first, Harder, is a poster of the type used by researchers at conferences to present their work, findings and its implications in summary form. Blood 3003 was the corresponding abstract. It was common ground that Harder would be read together with the all the Blood Abstracts, although it may have been Bayer's position, it was not clear, that this was the case only if the skilled team did not also read what were referred to as the "Kubitza Posters" because of the overlap of content. I will shortly consider the Kubitza Posters but it seems to me that the skilled team would have wanted to be as informed as possible. Since it is agreed that potentially the Blood Abstracts would have been included in the skilled team's reading, in my view they should be deemed to have been included irrespective of the Kubitza posters.
- The second, Perzborn, is an academic paper. Two of the references in Perzborn are Blood 3010 and Blood 3004 which are abstracts, concise summaries of other papers, published by ASH in its journal "Blood" in November 2003. Blood 3003 was on the same page in Blood as Blood 3004. By the time of the trial it had been agreed by the experts that Perzborn would have been read with all three Blood Abstracts as a single composite item of prior art.
Cross-references
- The authors of Blood 3004 and 3010, led by Dagmar Kubitza, also presented two posters which contained the work summarised in the two abstracts in expanded form. They were referred to as the Kubitza posters.
- Unusually it was the patentee, Bayer, which argued for the content of a cited item of prior art, in this case Harder, to be expanded by including with it the content of other prior art, here the Kubitza posters. The claimants submitted that the combination was misconceived in law, an impermissible mosaic.
- In this jurisdiction (unlike the tribunals of the European Patent Office and the courts of some European Patent Convention (EPC) States), inventive step is habitually assessed by reference to one item of prior art at a time. There is an exception where an item of prior art refers to another, in which case two or potentially a series of cross-referencing disclosures can be taken together, see Sharpe & Dohme Inc v Boots Pure Drug Co Ltd (1927) 44 RPC 367; (1928) 45 RPC 153, at 180. On one view, there is a further exception in that the skilled person may be taken to elucidate an item of prior art by referring to available information that he or she knows to exist, see for example Generics (UK) Ltd v Daiichi Pharmaceuticals Co Ltd [2009] EWCA Civ 646, at [25], but this will seldom amount to the combination of the total content of two documents.
- Harder expressly refers to the Kubitza posters and it was common ground that the skilled team reading Harder would have been motivated to read them. They were displayed in close proximity at the ASH meeting in San Diego in December 2003. On the other hand, their availability to the public was limited in that the posters were taken down and not subsequently displayed or otherwise published. They were no longer available at the priority date.
- The disclosure of information without any fetter of confidence makes it available to the public and part of the state of the art even if the disclosure is ephemeral, see AutoStore Technology AS v Ocado Group plc [2023] EWHC 716 (Pat) at [237] and Richter Gedeon Vegyeszeti Gyar RT v Generics (UK) Ltd [2016] EWCA Civ 410 at [14] to [16]. As of the date on which inventive step is to be assessed, the priority date, information once publicly disclosed but no longer available may be taken into account as prior art in the assessment. It follows from this and the agreed motivation to read the Kubitza posters that their content is to be considered and combined with that of Harder.
- Bayer wanted to take this a step further and said that in law Harder and the Kubitza posters are to be deemed read at the date when they were all actually available, namely the date of the ASH conference. Although the claimants challenged Bayer to explain why this was part of Bayer's pleaded case what difference it made I am not sure that the reason emerged. The usual and generally accepted hypothesis is that all prior art is considered by the skilled person as of the priority date of the patent in suit, see Generics (UK) Ltd v Daiichi Pharmaceutical Co Ltd [2008] EWHC 2413 (Pat), at [182]. The correctness of the date of assessment was not in dispute in Daiichi but I see no reason to depart from it, particularly since Bayer did not press the point save to say that it would be reserved for the Court of Appeal should it be necessary.
The issues in the case
- The claimants allege:
(a) The Patent lacks inventive step over:
(a) Harder and Blood 3003 plus, as I have found, the Kubitza posters; and/or
(b) Perzborn, together with Bloods 3004 and 3010, alternatively in addition together with Blood 3003.
(b) The specification of the Patent does not disclose the invention sufficiently clearly and completely for it to be performed by a skilled person because
(a) the claims contain no limitation as to dose; and
(b) claim 1 covers a range of thromboembolic disorders.
Both the foregoing allegations of insufficiency are based on issues of plausibility.
- The claimants' case on insufficiency is also pleaded as a ground for alleging that the inventions claimed are obvious in the AgrEvo sense, see EPO Technical Board of Appeal decision T-939/92 Triazoles/AgrEvo [1996] OJEPO 309. It was argued just by reference to the law on insufficiency on the basis that the outcome would be the same irrespective of the appropriate legal hook.
- In response to the allegations of insufficiency, Bayer has conditionally applied to amend the claims in two respects.
- The claimants contend that the Patent as proposed to be amended would be invalid on the ground of added matter.
BAY 59-7939
- Although Harder concerns rivaroxaban, it is not identified as such in the document. Instead the drug is referred to as BAY 59-7939, a designation used by Bayer. The claimants' case on obviousness depended on the skilled team at the priority date having access to the chemical structure of rivaroxaban.
- The claimants argued that at the priority date the skilled team would without undue difficulty have found out what the formula is by one or other of two routes.
- The first would have been simply to ask Bayer. By the priority date Bayer had publicly disclosed that BAY 59-7939 was rivaroxaban and had provided its structure. Consequently, the claimants submitted, the likelihood was that Bayer would have raised no objection to providing the information in response to an inquiry.
- The second route would have involved instructing an information specialist, an individual experienced in obtaining published scientific information. He or she would likely be in-house if the skilled team were part of a large pharmaceutical enterprise, otherwise an independent information specialist familiar with researching in the pharmaceutical field could have been commissioned to do the job.
Asking Bayer
- To deal with this aspect of the case, evidence was given on behalf of Bayer by Dr Frank Misselwitz. In January 2005 Dr Misselwitz was Global Clinical Strategist and Global Clinical Leader at Bayer. He was a co-author of the Harder poster, its corresponding abstract and the Kubitza posters.
- The claimants' expert clinician, Professor Hirsh, was asked to speculate about what Bayer would have done. As he said, this was not his area of expertise. His opinion on the subject could carry little weight. The evidence that mattered was that of Dr Misselwitz who had real knowledge of Bayer's likely behaviour at the relevant time.
- In his witness statement Dr Misselwitz said that there was a core team of 5-7 people, of whom he was one, which dealt with day-to-day matters regarding the BAY 59-7939 project. A request for information connected with a clinical investigation into BAY 59-7939 would have been passed to the core team and he would have been fully involved in the response. He said that save for an inquiry about the formula from a trusted academic institution, which if approved would have involved a formal agreement with confidentiality provisions, that information would not have been disclosed. A request made directly to the authors of Harder would have met with the same response.
- Dr Misselwitz further stated that Bayer twice disclosed the chemical structure for BAY 59-7939 before the priority date of the Patent, first during a presentation by a Bayer scientist at a meeting of the American Chemical Society in Philadelphia in August 2004 and then in the Perzborn paper, published on 26 January 2005. He accepted that Perzborn would have been carefully considered and approved by the relevant authorities in Bayer before publication and added that the information was also available in databases before January 2005.
- Despite this, in his witness statement he maintained that to the best of his recollection Bayer's approach to third party requests for information about BAY 59-7939 would not have changed after the August 2004 meeting because it would have put Bayer at a commercial disadvantage and would have caused a risk to Bayer's clinical development of the product.
- In cross-examination Dr Misselwitz could give no reason why disclosure of the formula would have had those adverse effects on Bayer when it was in the public domain. He was pressed several times on this. Although I think he attempted to avoid a clear answer, in the end he conceded that his evidence made sense if the inquiry had sought not just the chemical structure but also other critical information that would give a competitor an advantage in a research programme.
- It is possible that Dr Misselwitz's loyalty to Bayer and the high financial stakes associated with these proceedings may have led him to convince himself that Bayer's policy with regard to the disclosure of the chemical structure of BAY 59-7939 did not change after Bayer had made that information public, despite this making no obvious sense. My impression of his cross-examination was that he realised it made little sense and that probably Bayer's resistance to disclosure at that time would have been directed to other information potentially helpful to a competitor's research, not to the name and formula by themselves which Bayer had already willingly published.
- I think it is likely that a request for the generic name and chemical structure of BAY 59-7939 made to Bayer at the priority date would have been met with indifference from Bayer as to their disclosure and would have led to the supply of the information.
The information specialist
- The expert clinicians on both sides indicated that they would have wished to use an information specialist if the search for information was important to the skilled team. Bayer submitted that it may not be correct to assume that the search would have been seen as important. I disagree. The clinician and pharmacologist would not have known how important rivaroxaban would turn out to be, but there was a lot of interest in new antithrombotic drugs in January 2005, no doubt because pharmaceutical companies carrying out research in that area were well aware of the potential value of a successful product. It emerged that the hypothesised search for the information may have taken less than an hour and probably no more than three hours at the most. I think the skilled clinician and pharmacologist would have believed before the search that it would be time and money well spent.
- The claimants and Bayer each filed expert evidence from an information specialist. The claimants' expert was John Wickenden who has spent 45 years as an information scientist at Eli Lilly & Company Limited. Between 2000 and 2010 Mr Wickenden was Eli Lilly's representative for the Pharma Documentation Ring, an association representing the scientific departments of many leading pharmaceutical companies, including Bayer Healthcare.
- Bayer's expert was David Bawden who was formerly full-time, now part-time, Professor of Information Sciences at City, University of London. He is also an Honorary Lecturer at University College London.
- Both witnesses provided helpful evidence although Mr Wickenden's hands-on experience of the sort of research being hypothesized allowed him to give more assured and probably more realistic answers than some of those coming from the academic standpoint of Professor Bawden.
- The experts were agreed that the information specialist called upon by the skilled team would have had access to databases which contain information about drugs in development made available by the companies doing the work. They further agreed that a request for the identity of a drug from the code given by its developer was a common task.
- The hypothesis was that the information specialist would have been asked by the clinician and/or pharmacologist in the skilled team to find the name and chemical structure of BAY 59-7939. Mr Wickenden's evidence, which I accept, was that upon being given a task he was not paid to wonder whether he would succeed; he would set to and do his best to find it. It seems to me that the information specialist would have done likewise. The question is whether at the priority date he or she would have been likely to succeed.
- Mr Wickenden's best guess was that it would have taken him an hour to find the information if it was there. Professor Bawden said three hours. I think Mr Wickenden's experience made his estimate more reliable, but either way I take the view that the skilled specialist would have persevered for up to three hours and thereby to the point at which either the answer was found, or the conclusion would have been reached that the answer was not there to be found.
- The agreed first step would have been to use the Bayer code to search the CAS Registry, a database made available by a division of the American Chemical Society (ACS). It would have taken a few minutes. The answer would not have been found this way, but both experts thought it probable that one abstract would have emerged which would have suggested that it was quite likely that the chemical structure was disclosed at the ACS presentation in Philadelphia.
- Step two, again common ground, was to search the drug pipeline databases. Among the databases available in January 2005 were those hosted by the Prous Science. Prous' principal database, available to information specialists in the pharmaceutical field, is known as Prous Integrity. Prous also provides the industry with a database containing recent news of interest called Prous Science Daily Essentials.
- There was a conference in Philadelphia held by ACS from 22-26 August 2004 at which a Bayer scientist, Dr Susanne Roehrig, gave a presentation entitled "Discovery of the Novel Antithrombotic Agent BAY 59-7939, an Orally Active, Direct FXa Inhibitor". During the presentation it was stated that BAY 59-7939 was rivaroxaban and the chemical structure was disclosed. On the last day of the conference Prous Science included in their Daily Essentials resource a post headed "Discovery of BAY 59-7939 discussed at ACS meeting." The post referred to the drug's name, rivaroxaban, and set out its chemical structure.
- There was another piece in Prous Science Daily Essentials published on 28 December 2004 entitled "In vivo effects of bridging therapy with BAY 59-7939 in rats" which also disclosed the rivaroxaban name and its structure. They were further disclosed in the September 2004 editions of a Prous hard copy journal, the Prous Science Drug Data Report. The Drug Data Report was available online through the Prous Drug Data Report database.
- This database is dynamic in the sense that updates overwrite earlier pieces. It was therefore not possible to conduct a search in 2024 which would replicate a similar search in January 2005. However, Mr Wickenden's evidence was that it was overwhelmingly likely that information on Daily Essentials and the Drug Data Report would also have been on Prous' principal database, Prous Integrity. In cross-examination Professor Bawden accepted that it was likely.
- A key question therefore was whether the information specialist would have searched Prous Integrity at the priority date. The experts agreed that a search based on a code name was a simple one, employing a search string in the form of the code and variations on it.
- Professor Bawden said that he had not heard of Prous Integrity at the priority date. Yet by 2004 Prous felt comfortable in describing it as the world's largest commercial database available on bioactive compounds. A Bayer representative was reported to have said at a meeting in 2004 that the database was the fastest at being updated and was very strong on conference coverage. Conference coverage would be exactly what the skilled information scientist would have been interested in. A textbook entitled "Using the Pharmaceutical Literature" published in 2006 contained a chapter called "Competitive Intelligence" which it described as information that a pharmaceutical company would use to find out what its competitors are doing. There is a section headed "Drug Pipelines" which lists databases that provide a drug's generic name and structure. Six databases are listed in alphabetical order, one of which is Prous Integrity.
- These matters were put to Professor Bawden who accepted that Prous Integrity was one of the leading databases in January 2005. It is possible that Professor Bawden's academic background means that he has different priorities when keeping up with important new databases. Mr Wickenden was well aware of Prous Integrity but said that his employer, Eli Lilly, did not use it at the priority date because its content largely overlapped with another database called IDdb3 used by Lilly. Eli Lilly may not have been unique in this choice, but plenty of others in Mr Wickenden's place must have used Prous Integrity given it status as one of the leading databases.
- I find from that evidence that on the balance of likelihood the information specialist would have searched Prous Integrity, no doubt among other databases. The specialist would have found on Prous Integrity the generic name and chemical structure of BAY 59-7939, probably in less than an hour.
- The evidence from the experts showed that if the information specialist had not found the information on Prous Integrity, the search would have continued.
- The next step would have involved what were called Key Database Hosting Platforms. There were three and all contained the relevant information. The experts agreed that they would have been searched, but disagreed as to how the search would have been conducted. I will take this briefly. Mr Wickenden was confident that these platforms would have revealed the information sought. Professor Bawden's evidence initially was that the skilled specialist would have pursued blind alleys. However, in cross-examination he agreed that if his instructions were that the search was important, the specialist would have persevered and succeeded.
- I find that this third step in the search would have turned up the rivaroxaban name and chemical structure, within an hour or not much more probably.
- A skilled team reading prior art about BAY 59-7939 at the priority date would have wanted to know its generic name and chemical structure. The individuals in the team would have acquired that information either by asking Bayer or by requesting a data specialist to obtain it from published sources.
The law on inventive step
- Bayer submitted that the four-stage approach to inventive step in Pozzoli SpA v BDMO SA [2007] EWCA Civ 588 at [23] was to be applied. The claimants did not dissent.
- It is a multifactorial assessment, see Generics (UK) Ltd v H. Lundbeck A/S [2007] EWHC 1040 (Pat) at [72], approved in Conor Medsystems Inc v Angiotech Pharmaceuticals Inc [2008] UKHL 49 at [42]:
"The question of obviousness must be considered on the facts of each case. The court must consider the weight to be attached to any particular factor in the light of all the relevant circumstances. These may include such matters as the motive to find a solution to the problem the patent addresses, the number and extent of the possible avenues of research, the effort involved in pursuing them and the expectation of success."
- There was no dispute that the legal standard applicable to the obviousness or otherwise of a dosing claim was the same as any other, see Actavis Group PTC EHF v ICOS Corporation [2019] UKSC 15 at [42]. It was agreed that on the present facts "obvious to try with a reasonable expectation of success" was the relevant criterion. The Supreme Court has held that this criterion may be applied to the assessment of inventive step, where it is appropriate on the facts, see Actavis Group (cited above) at [65].
The issue on inventive step in summary
- The documents making up the groups which constituted the two cited items of prior art all came from Bayer. The skilled team reading each group at the priority date would have recognised that they all report the results of a phase I trials. As I have just found, the team would have found out that the trials concerned rivaroxaban.
- Bayer accepted in closing that the skilled team, having read the cited prior art, would not only have contemplated moving forward to conduct a phase II trial, there would have been an incentive to do so and they would have done. Having access to the chemical structure of rivaroxaban would have made that possible.
- It was also ultimately common ground that if (not accepted by Bayer) the skilled team opted for a phase II clinical trial in which rapid-release rivaroxaban was administered once daily, the drug would have been used as specified in claim 1 of the Patent. No independent inventive quality was ascribed to claim 2.
- In closing Bayer argued that this would have required the skilled team not only to go for a once daily dosing regimen, but also to select rapid-release rivaroxaban. Focus on the rapid-release aspect of claim 1 appeared to be an afterthought. A rapid-release drug is just a drug that has not been formulated into a sustained-release form. It was common ground that customarily in the life of a pharmaceutical, the rapid-release form is developed first with sustained-release formulations, if any, coming later. For reasons I will set out below, I find that the skilled team would have taken as read the notion that its phase II clinical trial would be conducted using rapid-release rivaroxaban.
- The only point in issue was whether the phase II trial would, at least in part, have involved a once daily dosing regimen. More exactly, whether the skilled team would have contemplated that such a regimen was worth trying, with a reasonable expectation of success. In fact, since it was common ground that such a regimen would have been contemplated, the issue resolves into whether there would have been a reasonable expectation of success.
Success
- Unlike a project in which a sustained formulation of a known rapid release drug is being investigated, a team developing a new drug has a blank sheet when feeling its way towards a dosing regimen which holds the promise of being both effective and safe. Once phase I trials have been completed, some guidance from the data becomes available but it is limited. The intended purpose of the drug is to treat a condition. The healthy volunteers in the phase I trial will not have that condition, so the phase I data can provide no direct indication as to the therapeutic efficacy of the drug. Nor can the phase I data provide direct evidence of the safety of the drug when administered to a patient suffering from the relevant condition, a patient whose response may be significantly less robust than that of a healthy volunteer.
- I have referred to the concept of a therapeutic window. Despite the clues from phase I data, a team conducting phase II trials for the first time cannot know where the therapeutic window lies in relation to patients having the relevant condition, or whether the window is narrow or wide. Nor can the team know which dosing regimens will result in the effect of the drug on patients falling within the therapeutic window.
- The difficulty is quite marked in relation to an anticoagulant such as rivaroxaban. As I have discussed, administering the new drug in a dosage that was too low posed a real risk of harm to patients in the study. Due to invasive surgery or for other reasons they would be vulnerable to clot formation. If such formation were not inhibited, or not sufficiently inhibited, a fatal thromboembolism in the brain or a lung or elsewhere could be the result. Equally, a high dose causing excessive anticoagulant activity could lead to bleeding, not just at the site of a wound but elsewhere, such as the brain, again leading to harm and even, potentially, the patient's death.
- The hypothetical team planning a phase II trial on the back of Bayer's phase I data would therefore have approached the trial with caution. If the team were to have considered trying a once daily dosing regimen, a reasonable expectation of success would have amounted to a reasonable expectation that such a regimen would not put the welfare of the patients in the phase II trial at unacceptable risk.
- During my reading for the trial, it occurred to me that one of the unusual features of the case was the basis on which an inventive step was apparently being claimed. The usual rationale behind a patentee's argument in favour of an inventive step based on the skilled person having no reasonable expectation of success is that the skilled person laboured under a conventional technical perception or prejudice regarding the relevant subject-matter. In consequence of this perception the skilled person would have had no reasonable expectation that a particular technical step would succeed. The inventor, being inventive, rejected the conventional prejudice, saw that there was in fact a reasonable expectation of success and made the invention.
- In the present case there would have been no such perceived technical barrier to the claimed invention on the part of the skilled team. The team would have known that it could conduct a phase II trial using a wide range of once-daily doses from one small enough to be sure that there is no excessive bleeding up to a dose large enough to be sure of an antithrombotic effect over 24 hours. Such a range would have been likely to find the therapeutic window somewhere in the lucky patients administered with one of the doses fitting the window.
- Rather, the skilled team here would have been held back (if that was the case) by an ethical barrier. Since the size and location of the therapeutic window was unknown, administration of a wide range of once-daily doses would have posed a real risk of including one or more that missed the window, causing harm to patients.
- Just reading the Patent one might at first suppose that Bayer's case on obviousness is that although the skilled team would not have taken the inventive step for ethical reasons, Bayer was prepared to put patients' lives at risk in a phase II trial and then claimed its reward in the form of the granted Patent.
- That cannot be exactly right because clinical trials may be performed only if approved by an external ethics committee. The relevant ethics committee must have approved Bayer's phase II proposal, so it is to be assumed that lives were not unacceptably put at risk.
- Presumably, where there is a reasonable expectation of technical success but patients are at risk of harm to an unacceptable degree in the event of a miscalculation as to dosing regimen, there is no reasonable expectation of success within the meaning given to that criterion in patent law. Neither side at the trial put its case in exactly that way but I think it is to be inferred.
- Given that the ethical question will be resolved by an ethics committee rather than the skilled team, the real criterion in the present case is whether the skilled team would have thought that it was worth applying to the committee for permission to conduct a phase II trial which included a once-daily regimen with a reasonable expectation that the committee would give permission, and whether it was likely that permission would be given.
- The case was not argued in that way. The criterion applied was just whether there would have been a reasonable expectation on the part of the skilled team that a once-daily dose falling would be both safe and effective. Of course, this and the two-part criterion just mentioned could on one view be seen to amount to the same thing.
- During exchanges with counsel I raised the potential ethical aspect to the case, which had not featured in their arguments, to make sure that there was no underlying current that I was missing. It led to Bayer filing a supplemental closing written argument which at the end summed up Bayer's position in this way:
"
it is not right to characterise the reason that once daily dosing is not obvious to the skilled team as being one of just safety concerns or prejudice of some kind, including an 'ethical prejudice'. Rather the principal reason is because the skilled team would not have had any reasonable expectation that dosing rivaroxaban only once daily in a rapid-release dosage form would be sufficient to maintain the antithrombotic cover needed to be efficacious."
- I doubt that can be right. A reasonable expectation of maintaining antithrombotic cover would very likely follow from a sufficiently high once daily dose. The real issue is one of balance, which up to then had appeared to be common ground. I will assume that it still is.
The invention story
- The discussion with counsel about a possible ethical dimension raised another feature of this litigation.
- The claimants submitted that in other jurisdictions in which parallel patents have been litigated, Bayer has cultivated an invention story. This, they argued, has proved influential in persuading courts elsewhere to find that the relevant patent is valid.
- I cannot say whether, and if so to what extent, judges in other courts were influenced by Bayer's invention story, although it is possible. I certainly think that if any of those courts wondered why it was that since Bayer felt able to include a once daily regimen in its phase II trial, the skilled team would not have done likewise, the invention story would have provided the perfect answer from Bayer's point of view.
- I will explain Bayer's invention story. Before the trial I was provided with a copy of the decision of the Board of Appeal of the European Patent Office in which the Patent was maintained as granted. I was also given copies of judgments in translation from the courts of three European Patent Convention (EPC) States. They are the Oslo District Court, the District Court of the Hague and the Patent and Market Court of the Stockholm District Court. Shortly after the trial I was sent the judgment of the Dutch language Enterprise Court in Brussels. There is forthcoming a trial before the German Federal Patent Court in Munich. As is usual, the court's summons for oral proceedings, scheduled for 29 July 2025, sets out the preliminary opinion of the court which gives a provisional finding on inventive step. Two judgments from common law jurisdictions were also provided before the trial, one from the Federal Court of Australia and the other from the Court of the Commissioner of Patents for the Republic of South Africa.
- Just recently I was sent the judgment dated 28 March 2024 of the 3rd Chamber of the Paris Tribunal and a second judgment of the District Court of the Hague, dated 27 March 2024 (in the first Hague judgment Sandoz sought to revoke the Dutch equivalent of the Patent, in the second it was Teva).
- Broadly, the current consensus is that the Patent in its equivalent forms in other jurisdictions does not lack an inventive step. That was the ruling of the EPO Board of Appeal and the first instance courts in Norway, the Netherlands (twice), Sweden, Belgium and Australia as well as being the preliminary opinion in Germany. The dissenting voices come from the judgments of the South African court and of the Paris Tribunal. Both the latter ruled that the relevant equivalent to the Patent is invalid for lack of inventive step over Harder plus the Kubitza posters.
- Although I have looked at these judgments and decisions, they can be of little direct assistance in my reaching a conclusion on the issues in this trial, even those applying what amounts to the same law as derived from the EPC. Aside from the risk of "group think" taking a hold, each court will have heard different evidence and argument presented according to local procedural rules which will have affected the conclusion of the court. See also Actavis Group PTC EHF v ICOS Corp [2019] UKSC 15, at [100]-[101].
- A point did emerge, though, which has some relevance to the way that Bayer may have approached this and equivalent litigation: the invention story.
- The Patent states that the invention is based on and is justified by a phase II trial in which dosing was largely twice daily but with one 30 mg once daily dosage included. The results reported in the Patent from this phase II trial showed that the once daily regimen was both safe and effective. That it was effective is said to have been surprising (paragraph [0012]). By implication the efficacy of the once daily dose, or possibly that it was both effective and safe, would also have been surprising to the skilled team at the priority date of the Patent.
- The Patent also implies that although Bayer did not expect once daily dosing to be safe and effective, they were nonetheless willing to include a once daily regimen. As I have said, it is to be assumed that the relevant independent ethics committee was satisfied that such a trial could be carried out despite Bayer's adverse expectation. It is this apparent contradiction which is explained away by the invention story.
- I can best take the story from the account given to the Brussels Enterprise Court. Dr Misselwitz was part of the team developing FXa inhibitors at Bayer at the relevant time. He provided written evidence to the Belgian court. Dr Misselwitz told the court that the phase II trials conducted by Bayer on rivaroxaban were in fact done in two stages. Initially, a phase II trial had been intended based on Bayer's phase I data (the data in the prior art documents as filed in this court). Dr Misselwitz's evidence describing this first phase II study is quoted in the Belgian court's judgment (the numbering is from Dr Misselwitz's witness statement):
"13.
Without knowing the therapeutic window, it would have been irresponsible following phase I trials if the development team had provided a dosing regimen for rivaroxaban in a rapid-release tablet taken once daily (i.e., once every 4 to 8 half-lives). No one proposed such a dosage regimen at this point in time.
"
- Dr Misselwitz continued:
"25. Study plans for clinical trials in patients are rigorously reviewed by the external steering committee of medical experts as well as by ethics committees and health authorities. Commercial interests must not be taken into account in this examination. In the selection of dosages and dosing regimens for phase II studies of anticoagulants, due to the seriousness of the disorder (thromboembolic complications are life-threatening) and the safety profile of anticoagulants (bleeding can also quickly become life-threatening), patient safety is the only consideration that counts, namely in terms of avoiding not only thromboembolisms but also bleeding complications.
26. For this reason, when planning the first phase II study for rivaroxaban on the basis of the phase I data available at the time, a once-daily dosing regimen did not come into consideration at all. As far as I recall, all those involved, both internally and externally, advocated that the first phase II study should be conducted solely with twice-daily (bid) or thrice-daily (tid) dosing regimens."
- According to this account, the first phase II trial went ahead, administering doses of 5 mg, 10 mg, 20 mg and 30 mg, all twice daily. The results gave Dr Misselwitz an idea:
"29. Over the course of the phase II studies mentioned above, we gradually found that rivaroxaban appeared effective and safe (i.e., effective at doses as low as 5 mg bid and still safe at 30 mg bid) over the entire dose range that was tested in this still relatively small study. In view of the expected narrow therapeutic range, this was a surprising result to me. The idea of requesting a change in the study protocol to also include a once-daily dosage came to me very late in the study because I interpreted the results of the 5 mg bid, 10 mg bid, 20 mg bid, and 30 mg bid in a special assessment not shared by other professionals."
- Despite this opposition to his idea of trying once-daily dosing in a phase II trial, Dr Misselwitz said that he was not discouraged. In consequence of his persistence, a notion took shape at Bayer to carry out an interaction study with enoxaparin, an approved anticoagulant. The potential risk of dosing below the therapeutic window could be alleviated by supplying enoxaparin as an emergency medication should it prove necessary. More from Dr Misselwitz:
"16. I was directly involved in the planning and design of the enoxaparin interaction study. This was neither a standardized study nor a study required by the regulatory authorities. It was developed and carried out by Bayer specifically on the basis of our fears regarding the risks associated with the development of new anticoagulants. This study was important because enoxaparin was the standard treatment at that time and we wanted to investigate whether, in the event of a failure of rivaroxaban, i.e., the formation of a thrombus, there was a way of saving the patient by injecting enoxaparin as an emergency medication. The study showed that this was possible and that enoxaparin could be injected as an emergency medication. The positive and confidential result of the enoxaparin interaction study, which was only known to the development team, provided a degree of certainty that in the event of potentially life threatening consequences in the case of a failure or an underdosing of rivaroxaban, an emergency medication was available that could also be administered in addition to rivaroxaban without complications."
- Dr Misselwitz told the Belgian Enterprise Court that this internal Bayer study was initially not sufficient to sway colleagues at Bayer or the external safety committee in favour of once daily dosing:
"30. Even with knowledge of these (not publicly available) study results, the inclusion of the once-daily 30-mg dosage in the existing phase II study was very controversial in the development team. In this context, I still clearly recall the numerous discussions we had both internally at Bayer and externally with several key opinion leaders and ethics committees who, in response to our original proposal, expressed strong safety concerns regarding the inclusion of a once-daily (od) dosage for rivaroxaban in phase II studies and suggested keeping to bid dosages instead.
31. Several people from the steering committee (in particular Prof. Haas and the head of the external steering committee, Prof. Eriksson) expressed great scepticism about my idea of administering rivaroxaban once a day in a rapid-release formulation given the relatively short half-life of rivaroxaban. I also faced great resistance within Bayer. For example, the then head of clinical development, Dr. Kemal Malik, the then head of the Bayer organization in the USA, Dr. Marc Thibonnier, and the head of Bayer's research and development in Japan, Dr. Erik Louvel, initially all opposed the idea of testing rivaroxaban on patients in a once-daily, rapid release formulation because in their opinion an anticoagulant with a half-life such as that of rivaroxaban cannot be used as a once-daily drug and that doing so would lead to risks and harm to the patients.
40. Among the experts there was thus serious reservations in November 2004 as to whether a once-daily dosage of rivaroxaban in the form of a rapid-release tablet would be safe and effective."
- However, the doubters were finally won over:
"41. The ODIXa-HIP-OD phase II study was finally approved by national health authorities, such as the FDA, and successfully conducted (see NIK7). In this context, however, I do recall that health authorities (I was directly involved in the discussions), especially in North America, also expressed concerns when we proposed the once-daily administration of rivaroxaban for phase II and III clinical trials.
42. The large number of clinically highly experienced individuals who had reservations underscores the fact that it was initially very difficult to overcome the scepticism and misgivings in the medical community that once-daily administration of rivaroxaban could be safe and effective and could warrant further testing in phase II and III trials."
- Belgium being a civil law jurisdiction, Dr Misselwitz was not cross-examined on any of this. I am not certain of the extent to which the Belgian procedure provides for disclosure and to the extent that it does, what categories of documents were likely to have been included, but I can see no sign of any documents having been provided by Bayer aside from Dr Misselwitz's statement concerning the invention story.
- Although Dr Misselwitz appeared as a witness in the trial in this court and was cross-examined, he only gave evidence about Bayer's likely response to a request for information about BAY 59-7939. He said nothing about the invention story reported in Brussels and elsewhere even though it would seem to be a very attractive account from Bayer's point of view.
- The claimants suggested that the reason for Bayer holding back from running the invention story in this court can be found in the judgment of Collis J in the South African proceedings. Collis J heard the trial relating to the validity of the South African equivalent of the Patent in the Court of the Commissioner of Patents for the Republic of South Africa.
- Before the South African trial Bayer had applied for and had been granted an interim interdict restraining the importation and sale by the defendants of products alleged to infringe the South African patent. Dr Misselwitz gave evidence at the interim interdict hearing which, I assume, was written evidence without cross-examination. That hearing was before Fourie J in the same court.
- Later came the full trial before Collis J, who delivered her judgment on 12 December 2023. Collis J noted that Dr Misselwitz and Dr Kubitza, both of Bayer, were among the authors of Harder. She also noted that Harder contained the assertion that some of the pharmacodynamic assays suggested BAY 59-7939 was suitable for a once daily dose regimen, what the judge called "the Harder observation". Collis J referred to an article by Drs Misselwitz and Kubitza published in 2016, sometime after their work on rivaroxaban had been completed but before thoughts of litigation had entered their minds:
"[316] In their 2016 retrospective article on how once-daily dosing came to be, Drs. Misselwitz and Kubitza stated the importance of the Harder observation to their decision to try once-daily dosing. No doubt this article was written candidly and unguardedly, at a time, certainly insofar as South Africa is concerned, when there was no suggestion of litigation."
- The invention story was part of Bayer's case before Collis J. So far as I can tell, it was not presented in full bloom I can find no reference to enoxaparin as a potential emergency anticoagulant providing reassurance about the safety of higher doses in a second phase II trial using a once daily regimen. However, the key part of the story, that the surprising finding of a relatively large therapeutic window came from a preliminary phase II trial, not the phase I study, and that surprising results in the preliminary trial led to Bayer's breakthrough thinking which in turn cleared the way to a second phase II trial with once daily dosing, was certainly presented. Collis J said:
"[321] What was put to Professor Greeff in cross-examination was that it was during the course of the phase II trials, that Bayer found that rivaroxaban was effective and safe across the entire 12-fold dose range tested (i.e. effective at doses as low as 2.5 mg bid and still safe in patients at total daily doses of up to 60 mg (30 mg bid)) and that this was a very surprising finding as no one had anticipated that rivaroxaban would have a relatively wide therapeutic window in patients. It was this knowledge that led Bayer to consider adding a 30 mg once daily regimen in the [second] phase II clinical trial."
- Collis J rejected Bayer's story:
"[322] No evidence was led to support the proposition put to Professor Greeff, although it is consistent with evidence which Dr Misselwitz gave in the interim interdict application. That evidence which Dr Misselwitz gave in the interim interdict application is at odds with what he stated in his 2016 retrospective article where he said that phase I data, not phase II data, led to the decision to test once-daily dosing. Perhaps this is why Dr Misselwitz was not called to testify.
[323] It is also at odds with the fact that ethical and regulatory approval was obtained for conducting tests in patients across this range of doses. If there had not been a reasonable prospect of these doses being both safe and effective in patients, they would never have been proposed or approved for the trials. As such, there can be no suggestion of being surprised that the doses performed as expected.
[324] The entire theory that a narrow therapeutic window was expected and that it was only discovered during phase II trials cannot withstand scrutiny. The objective facts point to the conclusion that Bayer knew it before phase II trials started. The Bayer experts persuaded the authorities and got regulatory approval for phase II trials across a wide therapeutic window (from 5mg twice daily to 40mg twice daily) before even the completion of phase I trials. They could only have got that from either the compound patent, the early phase I data or making an assumption from enoxaparin (the FXa inhibitor with the wide therapeutic window), or from a combination of these factors."
- Collis J commented on Dr Misselwitz's evidence in the interim application, which had contributed significantly to Fourie J's finding that the patent was prima facie valid:
"[330] In the interim interdict application, in response to Professor Greeff's conclusions about the phase I data, Dr Misselwitz stated three times that the data did not suggest suitability for once daily dosing. In the light of what is stated in the Harder posters, and his unguarded comments in his 2016 article, it is difficult to characterise this reply of Dr Misselwitz's as anything other than disingenuous. The plaintiffs were clearly, and understandably, not prepared to risk him as a witness."
- Later (at [340]) Collis J found expressly that Fourie J had been misled by the evidence given by Dr Misselwitz.
- I am not in a position to accept Collis J's findings or to reach any view on them. I note however that the invention story or a version of it was advanced before the judge and that she rejected it, reaching the conclusion that for the reasons she gave, Bayer's phase II study was based on their phase I data.
Common general knowledge
- The parties drew up a statement of agreed common general knowledge with an annex containing a list of CGK issues in dispute. I will leave some of the issues to be discussed in context but set out most of them here.
- PK and PD assays are informative about likely clinical efficacy but are not unerringly predictive. Predictive results could only be the product of phase II and III trials. The claimants sought to make something of Professor Crowther's evidence on this, taking him to have meant that phase II trials could only go ahead if supported by sufficiently predictive assays and that these could only come from phase II trials. They called it a "chicken and egg situation", in other words that Bayer was seeking to argue that no phase II trial could ever go ahead based only on data from a phase I trial. I did not understand Professor Crowther to be saying that. The go-ahead for a phase II trial can be justified by appropriately informative data from one or more phase I trials. If this was not what Professor Crowther meant, the clear bulk of the evidence proves it to be the case.
- There was a difference of emphasis regarding the relative importance that the skilled team would attach to half-life results from PK assays on the one hand and data from PD assays on the other. Bayer preferred the former. Part of their argument was that the most reliable half-life data for rivaroxaban available to the skilled team at the priority date was 3-4 or 4-6 hours. This would have suggested that once daily dosing would result in the concentration of the drug in a patient's plasma falling to zero well before the expiry of 24 hours thereby raising a significant risk of failing to prevent thromboembolism. The claimants preferred PD assays because, they said, these pointed to a persistence in anticoagulant effect 24 hours after a dose. I discuss this below.
- It was common ground between the experts that PA data showing the half-life of a drug is a starting point for selecting a dosing regimen and by implication for finding a regimen that will be safe and will afford an effective antithrombotic effect. It is information that will be taken into account along with PD data. The claimants made the point that it does not necessarily follow that an appropriate regimen will require dosing at a frequency of once every half-life, as proposed by Bayer by way of a rule of thumb. I accept that. The evidence showed that there are too many uncertainties regarding the effect of a new drug over time for there to be a universal rule of thumb across the board of dosing regimens for all known drugs. I think the cross-examination of Professor Wilkins (the claimants' expert on pharmacology) accurately reflected the totality of the evidence on how half-life data is used when going forward with a phase II trial:
"Q. 'Half-life' is the time it takes for the plasma concentration or the amount of drug ... to be reduced by 50%" citing Goodman and Gillmans. You would know that is a well-known student textbook?
A. Correct.
Q. And the statement is correct, is it not?
A. That is correct.
Q. It says: "When the drug substance is applied in no more than a therapeutically effective amount, which is usually preferred in order to minimize the exposure of the patient with that drug substance in order to avoid potential side effects, the drug must be given approximately every half [life]" citing Rowland and Tozer. You would accept that is a true statement?
A. I would not accept that is a true statement. I think it is one of the factors, but if we were just to dose on the basis of half-life, I think we would get into trouble with a number of drugs. I could cite steroids, ACE inhibitors and Beta blockers as examples of those. You would end up with overdosing basically.
Q. But qualification it is a starting point?
A. It is a starting point."
- Thus, dosing every half-life is not a universal guide to appropriate dosing regimens for all drugs. Moreover, there are guides to the appropriate regimen other than half-life. But half-life is an important contributory guide when beginning the search for a regime which is both safe and effective. This is consistent with what the authors of Rowland and Tozer (Clinical Pharmacokinetics: Concepts and Applications (3rd ed.)) indicate.
- The principal purpose of a phase II trial is to establish the optimum dose to take forward into a phase III study. This will involve a range of dose regimens in the hope of identifying an optimum regimen. There was disagreement about the extent to which a phase II trial would be designed with a range of doses so as better to identify the optimum dose. Professor Crowther in cross-examination accepted that this was the case, with the proviso that no dose would be used in the expectation that it would put patients at risk. Subject to that, it may be expected that some doses will turn out to be less effective than others. I accept that evidence.
- LMWHs were known to have a half-life ranging from 2-6 hours. At the priority date enoxaparin, an LMWH, was a known treatment for VTE. It is an FXa inhibitor although it does not target FXa directly, instead targeting antithrombin thereby catalysing the inactivation of FXa by antithrombin. Although LMWHs were known to exhibit no measurable anticoagulant effect beyond 12 hours and are sometimes administered twice daily, it was also part of the CGK at the priority date that they could also be effective in a once-daily dose.
- There was disagreement regarding the extent to which the known anticoagulant effect of LMWHs would have provided a guide to the likely effect of rivaroxaban and what that might mean for its therapeutic window. Professors Crowther and Hirsh were agreed that the mechanism of action of LMWHs differs from that of rivaroxaban. There are several potentially significant differences, including molecular structure, method of administration, target protein and method of FXa inhibition. Professor Hirsh believed that predictive parallels between the behaviour of LMWHs and rivaroxaban would have been drawn at the priority date, but he has a particularly high level of knowledge about LMWHs and did not identify any documents which would lead to the conclusion that such expected parallels formed part of the CGK at the priority date. I find that they did not. Professor Hirsh made a similar, though more reserved point about heparin and likewise I find that the skilled team would have made no predications about the therapeutic window of rivaroxaban based on heparin data because such data was not within the CGK.
- There was some suggestion from Professor Hirsh in his written evidence that it was known at the priority date that other FXa inhibitors had a wide therapeutic window, so that same may have been expected of rivaroxaban. In cross-examination he said that this was based on material made available at the ASH conference, held a month before the priority date. I am not satisfied that publication at the ASH conference led to the information becoming sufficiently widespread so as to attain the status of CGK.
- An issue arose as to the importance which the skilled team would have attached to the clinical and marketing desirability of a drug for the treatment of thromboembolic disorders, in a tablet form, that needs to be taken only once a day, as opposed to twice or more. Professor Hirsh said that it was highly desirable for reasons of patient adherence and convenience, and because of the associated marketing advantage. In cross-examination he conceded that it was not as important as seven other factors which he had identified in a paper authored by him, dating from 2005. Professor Crowther said that once-daily administration is desirable, he was not sure that it was highly desirable. He explained what he meant: it was never an endpoint in itself in that it did not override considerations of safety and efficiency. I accept Professor Crowther's evidence. The skilled team would have been aware of the clinical advantages of a once daily tablet over, say, a twice daily tablet. The team would have been part of a notional research department in a pharmaceutical company, not working in academic isolation. They would therefore have been fully aware of the potential financial advantage that would flow from marketing the first once daily tablet for thromboembolic disorders. However, both considerations would have been secondary to safety concerns when designing a phase II clinical trial.
The ribbon
- Professor Meibohm (Bayer's expert on pharmacology) stated that the skilled team conducting a phase II study with no knowledge of the size of the therapeutic window would aim for a relatively small peak-to-trough difference in the concentration of the drug in the plasma. This would increase the chance of the "ribbon" falling safely within the therapeutic window. The ribbon was the name given in argument to the wavy line graphically representing drug concentration in the plasm over time after steady state has been reached, oscillating between maxima and minima. Professor Crowther said something similar.
- Professor Meibohm illustrated the idea with the following graph, here showing a hypothetical therapeutic window, although before a phase II trial the skilled team would have no certain knowledge of where it would appear on the graph:
- In cross-examination Professor Meibohm said that doubling a dose and halving the frequency of administration would double the peak-to-trough distance, i.e. double the width of the ribbon. The converse was true: halving the dose and doubling the frequency of dose would half the width of the ribbon.
- A point to note about Professor Meibohm's graph is that the incline of those parts of the curve showing the rate of decline in concentration until re-administration of the drug is about the same for both regimens. That incline would be expected to reflect the ultimate half-life of the drug. Since regimens A and B are assumed to involve the same drug, it is to be expected that the inclines will be about the same. I think I can assume that Professor Meibohm intended this to reflect what happens in the real world, or at least that it is a safe approximation.
- Counsel for the claimants put another graph to Professor Meibohm in cross-examination, given the designation X5, which was part of a graph appearing in a section of Rowland and Tozer exhibited by Professor Wilkins in his report:
- In the legend to the original graph Rowland and Tozer say that dosing frequency controls the degree of drug accumulation. There may be more to Rowland and Tozer's graph than meets the eye. Regimen A has the same drug dose and presumably the same drug as regimen B, though administered at twice the frequency. Yet the rate of decline from peak concentration in regimen A is shown in X5 to be twice the rate shown for regimen B. To the uninformed mind that might imply a different half-life. But it was not discussed with Professor Meibohm and I take it no further.
- What Professor Meibohm said in relation to X5 was (a) the peak to trough variation in drug concentration is determined by the dose, (b) in X5 steady state has settled at a higher level because regimen A has double the rate of dosing (with the same dose) and (c) with the same dose at a different frequency you settle at a different steady state concentration.
- Professor Meibohm was cross-examined in some detail about his own graph, after he was taken to X5. It was not put to him that there was anything inaccurate or misleading about his graph and the answers he gave in relation to it confirmed his point that the skilled team contemplating a phase II trial with rivaroxaban, based on the phase I data in the prior art, would know that administering a relatively low dose at relatively high frequency would result in a narrower ribbon and a better chance of staying safely inside the therapeutic window wherever it turned out to be. He was not challenged on that assertion and I accept it.
- Nonetheless, this point about the ribbon does not decide the principal question: whether on the evidence any once daily dose contemplated by the skilled team would have been thought likely to be both safe and effective.
The first prior art citation: Harder plus the Kubitza posters
Harder
- The Harder poster begins:
"BAY 59-7939 is a selective, highly potent, direct FXa (FXa) inhibitor that is being developed for the prevention and treatment of thromboembolic disease. It has been shown to be well tolerated at single and multiple doses up to 30 mg, and is rapidly absorbed after oral administration, with a terminal half-life of 9-12 hours.
The aim of this study was to evaluate the effect of orally administered BAY 59-7939 on thrombin generation in healthy male volunteers."
- Under "Subjects and methods" the reader is told that the study was conducted on 12 healthy male subjects aged between 27 and 37. BAY 59-7939 was administered to eight of them while four had a placebo. The eight received a single 5 mg oral dose on day 1 and a single 30 mg oral dose on day 14, i.e. sufficiently later for the first dose to have been expelled from the body, or vice versa. Assessments of thrombin generation and platelet-inducing clotting were performed over 24 hours.
- Details of how the assessments were done are then given. There were three: a PITT assay, an ETP assay and a PICT assay.
- In the case of the ETP assay a graph is provided showing thrombin activity over time. The thrombin generation in patients receiving the drug started later and reached a much lower peak than in those receiving the placebo. These are the stated results:
"ETP (peak or AUC) was reduced significantly (compared with placebo profiles) by both the 5 mg and 30 mg doses of BAY 59-7939, with maximum effect at 2-4 hours. Inhibition of ETP-peak and ETP-AUC (induced by tissue factor or collagen) by 30 mg BAY 59-7939 was sustained over 12 hours (Figure 3)."
- This is the Figure 3 referred to, illustrating the effect of 5 mg and 30 mg doses of BAY 59-7939 on thrombin activity measured by ETP compared with a placebo. Graphs (a) and (b) show ETP peak values, (c) and (d) AUC. Graphs (a) and (c) show the values where ETP was induced by collagen, in graphs (b) and (d) it was induced by tissue factor.
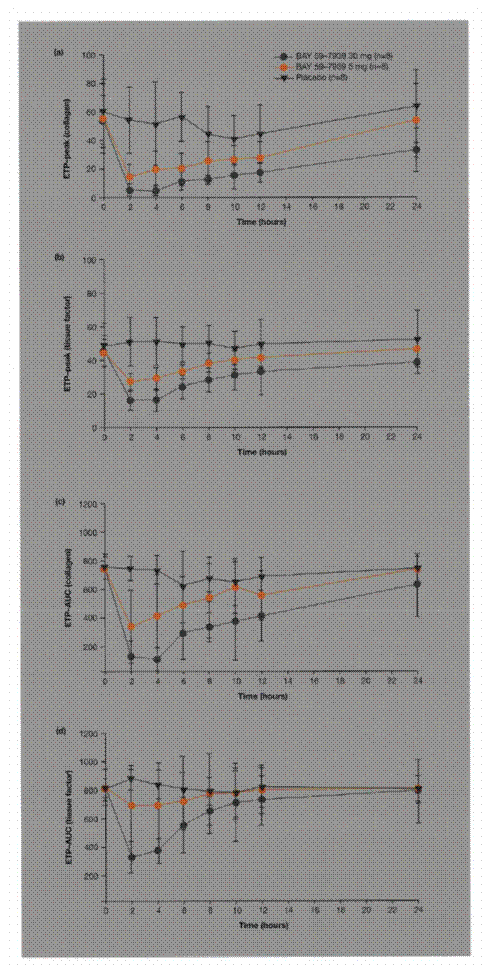
- The PITT results were discussed in Harder:
"PITT (Tc) was prolonged significantly (compared with placebo profiles) by the 30 mg dose of BAY 59-7939. Maximal prolongation of Tc was approximately 2-fold by 5 mg BAY 59-7939 and approximately 4-fold by the 30 mg dose (compared with placebo group), and was observed 2-4 hours after dose administration. The increase in PITT was sustained by 30 mg BAY 59-7939 over 12 hours (Figure 4a)."
- Also the PICT results:
"PICT was prolonged significantly (compared with placebo profiles) by both the 5 mg and 30 mg doses of BAY 59-7939. Maximal prolongation was approximately 2-fold by 5 mg BAY 59-7939 and 3-fold by the 30 mg dose, and was observed 2 hours after administration. PICT was prolonged over 12 hours after treatment with 5 mg and 30 mg BAY 59-7939 (Figure 4b)."
- Figure 4, like Figure 3, showed plots for the PITT and PICT results at 24 hours, and like Figure 3 both suggested some residual activity after 24 hours.
- Harder continues:
"In agreement with other phase I data [the Kubitza posters], FXa was inhibited dose dependently after administration of BAY 59-7939. Maximum inhibition was observed 2 hours (28% and 56% inhibition after treatment with 5 mg and 30 mg BAY 59-7939, respectively). FXa inhibition correlated closely with ETP, as demonstrated by the values of the ETP-peak."
- The findings are summarised by these conclusions at the end of the poster (excluding footnotes):
"● Orally administered BAY 59-7939 dose-dependently inhibited both intrinsic (collagen) and extrinsic (tissue factor) pathways of thrombin generation.
● The effect of BAY 59-7939 on thrombin generation was demonstrated in platelet-free assays and in PRP-based assays. In contrast, indirect (i.e. antithrombin III-dependent) FXa inhibitors obviously only inhibit FXa that is not protected by the platelet-prothrombinase complex.
● BAY 59-7939 not only inhibited the lag time of thrombin generation (PITT-T�c), but also had a profound effect on both the maximum extent of thrombin generation (ETP-peak) and the total amount of generated thrombin (ETP-AUC). This observation suggests an additional feature of BAY 59-7939, because weaker FXa inhibitors may only prolong lag-time without affecting the total amount of thrombin generation.
● Some parameters (e.g. ETP-peak) indicate a long-lasting pharmacodynamic effect of BAY 59-7939, which suggests suitability for a once-daily dosing regimen.
● The effects of oral BAY 59-7939 on intrinsic and extrinsic thrombin generation and PICT, which are mediated by direct inhibition of FXa, indicate that BAY 59-7939 is a promising anticoagulant that merits further clinical investigation."
The Kubitza posters
- There are two Kubitza posters, both in a format fairly similar to that of the Harder poster but with more detail. The first is entitled "Single dose escalation study investigating the pharmacodynamics, safety and pharmacokinetics of BAY 59-7939 an oral, direct FXa inhibitor in healthy male subjects". The title of the second is the same, save that it is a "Multiple dose escalation study". As their titles imply, they differ in that the first involved the successive daily administration of the drug or a placebo in single ascending doses (SAD) over the period of the study. For the second, the volunteers were given multiple ascending doses (MAD) or a placebo. They were referred to in the evidence as the SAD and MAD studies.
- The SAD study was carried out on 103 healthy men under fasting conditions. Some received BAY 59-7939 in 9 steps, being successive doses of 1.25, 5, 10, 15, 20, 30, 40, 60 and 80 mg tablets. BAY 59-7939 was administered to others in two oral solutions of 5 and then 10 mg. Yet others received placebos.
- Pharmacodynamic and pharmacokinetic tests of the volunteers' blood were carried out:
"● Pharmacodynamic effects were evaluated using FXa activity, prothrombin time (PT), activated partial thromboplastin time (aPTT) and HepTest; selectivity was assessed by measuring Factor IIa (FIIa) and antithrombin III activity.
● The pharmacokinetic parameters measured included area under the plasma concentration-time curve from zero to infinity (AUC) maximum drug concentration in plasma (Cmax), and half-life associated with terminal slope (t1/2)."
- The results from the pharmacodynamics were given and in the case of FXa activity this included duration (here omitting the references to figures):
"Pharmacodynamics
All pharmacodynamic parameters had similar dose-dependent time-response curves, although the magnitude of the curves varied depending on the parameter. Overall, pharmacodynamic parameters were slightly more affected after administration of oral solution than after tablet administration.
FXa activity and specificity
Median FXa inhibition ranged from 20% for 5 mg tablets to 61% for 80 mg tablets. The maximum inhibitory effect on FXa activity was observed 1-4 hours after tablet administration, and returned to the normal range (0.7-1.2 U/mL) within 24 hours for doses up to 40 mg. FXa activity remained elevated beyond 24 hours for the 60 mg and 80mg doses. BAY 59-7939 was specific for FXa and did not affect FIIa or antithrombin III."
- As Professor Meibohm observed, there is a typo in the third sentence. BAY 59-7939 is an inhibitor of FXa. It should read "Inhibition of FXa remained elevated beyond 24 hours
" etc. This observation was supported by the data shown graphically in Figure 1. It is difficult to decipher Figure 1 as reproduced here but this gives an impression:
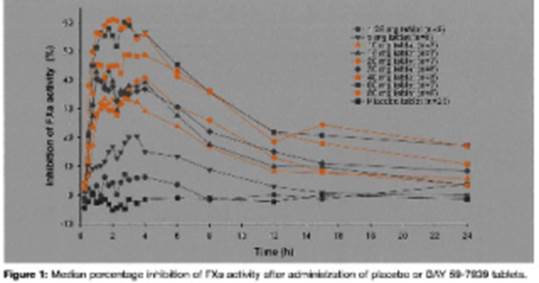
- Professors Hirsh and Wilkins said that the skilled team would have noticed from the Figure that inhibition of FXa is shown to remain elevated above the baseline at 24 hours for the 30 and 40 mg doses as well as the 60 and 80 mg doses. They were not challenged on this. The same appears to be true of the PT assay.
- Professor Meibohm disagreed in his first report. He said that for doses up to 40 mg FXa inhibition converges with the placebo. To my eye, Professors Hirsh and Wilkins were plainly correct, with particular reference to a dose of 30 mg. In his second report, I think Professor Meibohm made a careful though only implied concession that Professors Hirsh and Wilkins were right, adding that the skilled pharmacologist would not know whether any particular level of FXa inhibition has therapeutic effect. This latter point is of course true, though I think not helpful. Taken to its logical conclusion none of the PD data reporting FXa inhibition was of much value because it would not have been known whether any particular level of inhibition had therapeutic effect. Yet plainly those in the field regarded such data as the best PD data they had to work with and, more importantly, they believed that it was of sufficient indicative value to be taken seriously.
- The pharmacokinetic results in the SAD study included an estimate of the half-life of BAY 59-7939 in the volunteers' plasma when administered as an oral solution (no half-life estimation for tablets is given):
"Plasma concentration-time profiles showed rapid absorption after administration of the solution. Maximal plasma concentrations were achieved after 30 minutes and t1/2 was estimated to be 3-4 hours."
- FXa inhibition was plotted against plasma concentration, showing a close correlation at higher plasma concentrations. At low plasma concentrations, small increases in plasma concentration were shown to cause a relatively large increase in FXa inhibition. PT results, prothrombin time, were also plotted against plasma concentrations, showing a consistent close correlation.
- The poster stated in its conclusions that there was correlation between PD effects and plasma concentrations. The poster also concluded that further investigation of BAY 59-7939 was warranted.
- The MAD study was with 64 subjects, of whom 61 also took part in pharmacokinetic analyses. The volunteers were each randomly assigned to active treatment or administration of a placebo. Treatment was in 6 steps, the first on day 1 and then on days 4-8. For those receiving BAY 59-7939, the successive daily dosing regimens were: 5 mg once, 5 mg twice, 5 mg three times and then 10 mg, 20 mg and 30 mg in each case twice daily. Pharmacodynamic (FXa activity, PT, APTT and HepTest) and pharmacokinetic tests were conducted along similar lines to those in the SAD study.
- Professor Hirsh noted that the inhibition of FXa was slightly above baseline after 24 hours with the twice-daily administration of the highest dose used (30 mg). FXa inhibition also remained slightly above the baseline 24 hours after the final dose. This correlated with plasma concentrations of the drug, which also remained above the baseline 24 hours after the final dose. Similar sustained effects could be observed in relation to PT. Professor Hirsh added this:
"The Skilled Clinician would consider that these data were consistent with the prolonged pharmacodynamic effects seen in the 30 mg does in the [SAD study] and with the sustained effects at 24 hours in some of the thrombin generation assays seen at this dose in the Harder Poster."
- The poster itself reported:
"● Cmax was reached 2.5-4 hours after administration, and the [half-life] of the 5 mg dose was 5.4 hours.
● For the 10 mg and 30 mg doses, the mean [half-life] was 5.8 hours, and for the 20 mg dose it was 3.7 hours."
- As with the SAD study, FXa inhibition and PT results were plotted against plasma concentration. The results were similar.
- These were the bullet point conclusions:
"● BAY 59-7939 was safe and well tolerated after multiple-dose administration at all the doses tested without signs or symptoms of bleeding. BAY 59-7939 inhibited FXa activity and dose-dependently affected the pharmacodynamic parameters PT, aPTT and HepTest.
● Predictable dose-dependent pharmacodynamics and pharmacokinetics were demonstrated, and indicated that BAY 59-7939 is suitable for twice-daily administration up to 30 mg.
● Close correlation existed between the prolongation of the PT and plasma concentrations
● The good safety profile, selective inhibition of FXa and promising pharmacokinetic and pharmacodynamic profiles indicate that further investigation of BAY 59-7939 is warranted."
The differences between the prior art and the claimed invention
- In closing Bayer identified four differences between the phase I data of Harder and the Kubitza posters on the one hand and the phase II use of rivaroxaban covered by the claims of the Patent:
(1) The lack of identification of BAY 59-7939.
(2) The absence of any mention of a rapid release form.
(3) Treatment of patients vulnerable to thromboembolism, as opposed to healthy phase I patients.
(4) Administration once daily for at least five consecutive days.
- I have found that the skilled team would become aware that BAY 59-7939 is rivaroxaban.
- I have already mentioned that Bayer said nothing about relying on the rapid release form until closing submissions. Nonetheless, since Bayer took the point at the last minute I will address it.
- Both Professors Meibohm and Crowther said that the prior art did not disclose a rapid release formulation of rivaroxaban to prevent or treat thromboembolic disorders administered no more than once daily for at least five consecutive days. But neither singled out the rapid release feature as being of any significance by itself. Since Bayer's opening submissions did not either, it was not surprising that the claimants' cross-examination was not much directed to the point, though Professor Meibohm spoke of the skilled team considering phase II dosage using rapid-release rivaroxaban (not sustained release) because that was what had been used in the Kubitza posters.
- It was put to Professor Wilkins that if the skilled team wanted to minimise peak-to-trough fluctuations, using a controlled-release formulation would be an appropriate step. Professor Wilkins said:
"A.
if you wanted to minimise fluctuations because you knew about the therapeutic window, in other words your freedom to operate between a safe dose and an effective dose, then that would be an appropriate step."
- He went on to say that the skilled team would never know what the therapeutic window is at this stage in drug development.
- Professor Wilkins' answer that I have quoted speaks of knowing the therapeutic window. I think he probably meant that once the window has been identified, one way to go would be to target it using a sustained-release formulation. That would not apply to a phase II trial done before the size and location of the window is known.
- I am satisfied that the skilled team contemplating a phase II trial based on the phase I data of the prior art would think first and probably only about using rapid-release rivaroxaban.
- Turning to Bayer's third difference, the undisputed evidence was that the skilled team would have it well in mind that the patients in a phase II trial would be more vulnerable than the healthy patients at both ends of the spectrum: more liable to bleeding if the drug level is too high and more likely to develop thromboembolism if it is too low. It is a reason why considerable caution is required when designing a phase II trial. Nothing was said in evidence that gave the difference between the nature of the patients in the two types of trial any greater significance than this. The evidence from phase I is not predictive in the sense of giving certainty as to what will happen in a phase II trial, in part because of the difference in the condition of the patients, but it provides important and necessary guidance.
- As to the fourth difference, the experts attached no significance to the five consecutive days. The criterion of once daily treatment only makes sense if it happens for more than one day and the evidence did not indicate that five days was anything more than an obvious and probably arbitrary period to select.
- The only real issue in the case on inventive step was whether it was obvious for the skilled team to conduct a phase II trial which included once daily dosing having read Harder and the Kubitza posters.
The experts' evidence on once daily administration
- Harder says that assessments of thrombin generation and platelet-induced clotting were performed over 24 hours, including with a 30 mg dose. It states that the effect of the 30 mg dose was sustained in each case over 12 hours, as was the case in the PICT assay, but makes no mention of the effect after 24 hours. The graphs show plots for up to 24 hours after administration, suggesting in 3 out of 4 cases that there was residual activity after 24 hours, although the last plots for 24 hours have error bars.
- Professor Meibohm thought that because the data was based on only 8 patients and the lack of plots between 12 and 24 hours after administration, the data was scientifically weak. This was consistent with the authors being prepared only to say that the effect of the 30 mg dose was sustained over 12 hours. Professor Crowther agreed, adding that the overlapping error bars meant that it was not possible to tell the degree to which there was an anticoagulant effect after 24 hours.
- Professor Hirsh accepted that the authors of Harder had not said that there was a sustained effect beyond 12 hours but pointed out that they did say that some assays suggested suitability for 24 hour dosing. Professor Wilkins expressed the view that if the skilled team were to dose at 12 hours, and re-dose at 12 hours, the ETP-peak assay suggested that thrombin generation would be inhibited for 24 hours. This was not clear. He may I think have meant that having climbed to steady state, thereafter dosing every 24 hours would prove sufficient, but he was not asked to clarify.
- The balance of the experts' evidence was that only limited weight one way or the other could be attached to the Harder data if considered in isolation.
- However, there remains the authors' comment that some parameters suggested a suitability for once-daily dosing. This seems to have been made with the data from the Kubitza posters also in mind because they expressly stated that the FXa inhibition data was in agreement with the data in the Kubitza posters. Harder and the Kubitza posters have to be taken together.
- The experts' overall conclusions on Harder and the Kubitza posters combined were as follows.
- Professor Meibohm said that the skilled pharmacologist would defer to the skilled clinician in reaching a decision whether to take BAY 59-7939 forward into a phase II study at all based on the phase I data available.
- If the skilled clinician approved moving forward to a phase II study, having read Harder and the Kubitza posters, the skilled pharmacologist would consider 20 mg and 30 mg given twice daily because the phase I study had shown that up to 30 mg twice daily was safe and well tolerated. A dose at least twice daily would be thought necessary based on the data in the Kubitza posters and the need to avoid excessive peak-to-trough fluctuations in the concentration of the drug in plasma.
- Professor Crowther emphasised more than once that there was no established correlation between the results generated by FXa inhibition, PT or APTT assays on the one hand and therapeutic efficacy or safety on the other. He said that the information in Harder was insufficient for the skilled clinician even to consider taking BAY 59-7939 forward to a phase II trial. The further information presented in the Kubitza posters was helpful since they provide results from more conventional phase I studies. However, the posters do not provide evidence of how statistically robust the data shown is and since posters would not have been subject to peer review the conclusions would have been viewed with significant scepticism. The skilled clinician would have thought that the drug was suitable for further development, but there was not enough detail in the combined disclosure to recommend any dosage regimens that would be expected to be sufficiently safe and effective to test in patients in a phase II trial.
- If BAY 59-7939 were to be progressed to phase II trials, contrary to Professor Crowther's primary view, he said that the decision as to which dosage regimens to test would be led by pharmacokinetic analyses performed by the skilled pharmacologist having regard to the skilled clinician's requirements. The skilled clinician would have expected it to be necessary to administer the drug at least twice daily to ensure sufficient concentration in the plasma over a 24 hour period. No weight would have been given to the assertion in Harder that BAY 59-7939 has a half-life of 9 to 12 hours because no data is given in support and it differs from the half-life values reported in the Kubitza posters.
- Professor Wilkins' opinion based on the phase I data of Harder and the Kubitza posters was that the skilled pharmacologist would want to go forward with phase II studies in respect of at least the following: 5 mg twice daily, 15 mg twice daily and 30 mg once daily. With regard to the last proposal, a dose of up to 80 mg was shown to be tolerated and the MAD poster demonstrated that a regime of 30 mg twice daily was safe. The PD data indicated a sustained effect with 30 mg after 24 hours, suggesting that a once daily 30 mg dose would be appropriate. There would be a reasonable expectation of success in terms of both safety and efficacy. The graphs for the PICT and PITT assays support the statement in Harder that BAY 59-7939 may be suitable for once daily dosing. The Kubitza posters would provide encouragement for this proposal, particularly since the pharmacodynamic data in the MAD poster did not show any sign of drug accumulation beyond steady state at a dose of 30 mg. His proposed dosing regimens would be subject to discussion with the skilled clinician.
- Professor Hirsh thought that the skilled clinician would take seriously the report in Harder that BAY 59-7939 indicates a long-lasting pharmacodynamic effect, in some assays for as long as 24 hours. The graphs in the Kubitza SAD poster indicated that inhibition remained well above the baseline 24 hours after administration of a 30 mg dose. The MAD poster indicated the same. Dosing at 30 mg would be an upper limit. but the skilled clinician would want to use that dose once daily in a phase II trial. The skilled clinician would have agreed with the assertion in Harder that some of the parameters suggest the suitability of BAY 59-7939 for once daily dosing, in particular ETP peak in the collagen assay and, to a lesser extent, ETP peak in the tissue factor assay and in the PICT assay.
- Professor Wilkins and Professor Hirsh stated their views about including a once daily 30 mg dose before seeing the Patent.
- None of the experts significantly changed his evidence in cross-examination. In their closing submissions Bayer quoted extended passages from the cross-examinations at some length to suggest that both Professors Hirsh and Wilkins retracted what they had said in writing. That is not my reading of the oral evidence. With regard to Professor Hirsh, his evidence in the transcript of the proceedings at Day 3, pages 291 to 294 was quoted in extenso by Bayer in their written closing submissions. It was followed by a submission that this passage showed that Professor Hirsh had agreed with Professor Meibohm's written evidence and that this was "really an end of the matter". If Bayer meant that it foreclosed the need for further debate about inventive step, I disagree. Parts of Professor Meibohm's report were put to Professor Hirsh, who did agree with them, but they were matters not in dispute. Professor Hirsh made his point about the predictive value of heparin and LMWH and as I have found, this carries no real weight. At the end of the passage counsel quoted to Professor Hirsh Professor Meibohm's assertion that the skilled team would have been of the view that BAY 59-7939 would have to be administered at least twice daily or more frequently. Counsel then said to the witness "
we see that you disagree and I think we found out exactly where the issues are joined." Dr Hirsh answered "Yes". If anything, this passage demonstrates that Dr Hirsh maintained his disagreement with Professor Meibohm and Professor Crowther.
- As to Professor Wilkins, in their closing argument Bayer referred to a section in the transcript of his cross-examination. There it was put to him that the Kubitza posters suggested that twice daily dosing was required. Professor Wilkins readily agreed. The Kubitza MAD poster states that "BAY 59-7939 is suitable for twice daily dosing". Although the question put to Professor Wilkins included the word "required", I did not have the impression that he intended his answer to be an abandonment of all the principal views expressed in his reports.
- There was one relevant concession that Professors Hirsh and Wilkins both made in their respective cross-examinations. It was that the plots beyond 12 hours in Harder, with their error bars, may not have statistical significance.
Once daily administration the headline arguments
- The claimants emphasised that the skilled team would not require phase I clinical data to provide certainty as to the safety and efficacy of once daily dosing, only that the data indicated that patients in a phase II trial would not be put to an unacceptable risk. The claimants submitted that a phase II study including a once daily regimen, based on the phase I data of Harder and the Kubitza posters, would not have created an unacceptable risk for the following headline reasons.
- First, the authors of Harder, two clinicians stated by Professor Crowther to be of international renown and scientists from a world-leading pharmaceutical company, told the reader expressly that the data disclosed suggested the suitability of BAY 59-7939 for once-daily dosing.
- Secondly, Blood 3003 stated in terms that the 30 mg dose achieved an anticoagulant effect for 24 hours.
- Thirdly, once daily dosing was known to be highly desirable from both a clinical and financial perspective.
- Fourthly, the graphs in the Kubitza posters confirmed sustained anticoagulant activity after 24 hours.
- Bayer emphasized that the skilled team's approach to phase II trials would above all else be one of caution. This was all the more so because of the known and quite recently published phase II trials using razaxaban. Lives were potentially at risk. Based on the phase I data of Harder and the Kubitza posters the skilled team would have had no reasonable expectation that a once daily dose would be both safe and effective for patients in a phase II study principally for the following reasons.
- First, the half-life reported in Harder would have been regarded as a much less reliable figure than those stated in the Kubitza posters. The half-life of 9-12 hours reported in Harder is not backed up by any PK data. The shorter half-lives stated in the Kubitza posters have the backing of data. Also, Harder was a small study of 12 subjects; the studies in the Kubitza posters used 108 and 66 subjects. The half-life of 3.7-5.8 hours reported in the Kubitza posters indicated that dosing at least twice a day would be necessary.
- Secondly, the Kubitza posters both reported a correlation between plasma concentration and PD results. The skilled team would conclude that the PD results must conform with a half-life of 3.5-5.8 hours, which would rule out once daily dosing.
- Thirdly, the skilled team would have regarded the PD assays in Harder as having no statistical relevance and those in the Kubitza posters as having no greater relevance.
- Fourthly, the statement in Harder that PD parameters suggested suitability for a once daily regimen would have been dismissed as unsupported and of no weight.
Discussion
- The key criterion in the assessment of inventive step in the present case, at least as it would be in the real world, is whether the skilled team would have sought the approval of the relevant external ethics committee for a phase II study including a once daily regimen, based on the phase I data contained in Harder and the Kubitza posters, and whether the committee would have given its approval. The difficulty is that there was no direct evidence as to how such a committee would go about its decision, in particular on a scale between complete intolerance of anything with a chance of causing harm to a patient down to something very much less stringent than that.
- The parties argued the case according to whether the skilled team would have thought that such a regimen was worth trying in a phase II trial with a reasonable expectation of success, where success would mean that such a regimen would be both sufficiently safe and sufficiently effective in a phase II study.
- In the real world a team's concern would be whether a proposed phase II study would pass muster with the ethics committee. The team's assessment of the appropriate degree of caution would presumably reflect that. I think the only approach that makes sense is to assume that the appropriate degree of caution on the part of the skilled team would have been the same as that applied by the notional ethics committee. But it takes one back to the same difficulty.
- I think two matters which were in evidence provided an idea of what this would mean. First, data from a phase I trial can never be predictive of what may happen in a phase II trial. In the case of a study involving a drug for treating thromboembolic disorders a phase I trial of the type disclosed in Harder and the Kubitza posters can only test anticoagulant activity ex vivo. That is not the same thing as, and need not necessarily correlate closely with, antithrombotic activity. The results of the trial do not strictly prove anything with regard to antithrombotic activity. A highly risk averse approach would mean that phase II trials would seldom if ever be conducted. That would not be in the public interest and is clearly not the approach adopted in the real world.
- Secondly and related to this, the examples of trials with razaxaban being abandoned because of severe excessive bleeding in patients and idraparinux causing bleeding in phase II trials which proved fatal (see the section on CGK above) establish that some degree of risk of patient morbidity and possibly even of patient mortality is inevitable and therefore necessarily tolerated, albeit of course with appropriate efforts to minimise the risk. The phase II trials of both razaxaban and idraparinux must have received approval from an independent ethics committee. The public interest in having new and valuable drugs progress through the pipeline of clinical testing to become available to patients has to be balanced against the risks to individuals in the clinical trials, including phase II trials.
- With that in mind, I turn to the question whether the skilled team of the present case would have considered that there was a reasonable expectation that a once daily regimen, using any chosen dose, would be sufficiently safe by avoiding an unacceptable risk of bleeding and also sufficiently effective by avoiding an unacceptable risk of thromboembolism during the course of a phase II trial.
- The expert clinicians and pharmacologists on each side remained apart on this key issue.
- A problem I have with the evidence of Professors Crowther and Meibohm taken together is that theirs was primarily a counsel of despair. Taken jointly, as it should be, their evidence was that the data from Harder and the Kubitza posters would have led the skilled team to abandon the idea of a phase II trial altogether. Of course, the quality of phase I data must vary from study to study and in theory this data could have been of a quality below the threshold required to go ahead with any sort of phase II study. But even Bayer's counsel in oral submissions was not prepared to go that far. He accepted that the skilled team would not only have contemplated a phase II trial, it would have conducted one. The only issue was whether it would have included a once daily regimen. This does not mean that the evidence of Professors Crowther and Meibohm was of no value it was both valuable and helpful and they had a fallback position on the assumption, of which they disapproved, that the skilled team would have conducted a phase II trial, evidence which I have taken into account. But it seemed to me that there was to some extent a determination to be negative.
- As I have found, the skilled team in this case would have had been aware of the clinical advantages of a once daily tablet and the financial potential of marketing the first available once daily tablet for the treatment and prevention of thromboembolic disorders should that prove possible. It would certainly have been secondary to their safety concerns. But even if that awareness would not have been at the forefront of the skilled team's thinking before reading the prior art, where the prior art raised the possibility of a phase II trial including a once daily regimen, that possibility would have been given serious consideration.
- It follows that the skilled team would have found the statement in Harder that some parameters investigated in the document suggested the "suitability [of orally administered BAY 59-7939] for a once-daily dosing regimen" both striking and encouraging. The authors of Harder were either, like Dr Harder himself, distinguished clinicians of apparently international renown or, like Dr Misselwitz, senior figures in Bayer, very much a leader in the field. The skilled team would in my view have considered carefully why these authors had been prepared to put their names to that statement. Further, Blood 3003 (the abstract corresponding to Harder with the same authors) said that some assays pointed to an anticoagulant effect lasting 24 hours after a single 30 mg dose.
- The skilled team would have been likely to consider first the reported evidence on the half-life of BAY 59-7939. Harder reported a figure of 9-12 hours, supportive of the idea of once daily dosing. But as Professors Crowther and Meibohm pointed out, there was no PK data to support it and the study involved only 8 participants treated with BAY 59-7939. The numbers of volunteers in both the Kubitza posters were much larger and there was supporting data. The figures of 3.7 and 5.8 hours in the two Kubitza posters would have been seen as more reliable.
- The authors of Harder must have had the data in the Kubitza posters in mind when the statement regarding suitability for once daily dosing was made. Harder refers to the data of the Kubitza posters as being supporting of at least some of the PD data reported in Harder. And the authors would probably not have pointed out that some data supported once daily dosing if such data were to be dismissed as of no value because of other data in Harder and/or the Kubitza posters.
- The skilled team would therefore have turned to the PD data. Harder stated that the ETP results showed that the inhibitory effect of BAY 59-7939 was sustained over 12 hours. As Professor Hirsh said, the graphical evidence gave some limited support for a sustained anticoagulant effect over 24 hours, although the authors had not seen fit to state this and the error bars meant that the evidence may not have had statistical significance.
- The larger and therefore more reliable studies in the two Kubitza posters suggested in their graphs that anticoagulant effect as measure by some parameters was significantly sustained 24 hours after administration in the case of 30, 40 and 60 mg doses. On the other hand these posters also concluded that anticoagulant activity according to a PD assay was closely correlated to plasma concentrations. The apparent contradiction, if it was seen as such by the experts, was not directly confronted. I note that the graphs in the Kubitza posters show that at low plasma concentrations of BAY 59-7939 there is not a close correlation between concentration and FXa inhibition. Even at zero concentration prothrombin time did not fall to zero. It could be that at the low plasma levels expected 24 hours after administration anticoagulant activity would no longer have been expected to correlate with plasma levels of BAY 59-7939, but the detail of the graphs was not addressed. As against that, it was resolved in the evidence of Professors Hirsh and Wilkins, and conceded by Professor Meibohm, is that the SAD Kubitza study shows graphical evidence of anticoagulant activity 24 hours after administration, including 24 hours after administration of a 30 mg dose.
- Taking all the evidence together, I think that the skilled team would have believed that it was reasonable for the authors of Harder to say that there was data in Harder and the Kubitza posters which suggested that BAY 59-7939 was suitable for orally administered once daily administration. That left the question whether the data indicated that there was a dose which would be both safe and effective.
- The experts were agreed that the data indicated a wide tolerance of BAY 59-7939 in healthy patients, up to a dose of 80 mg. A once-daily dose of 60 or 80 mg would be the most likely to prove effective in treating and preventing thrombi in phase II patients, though high doses of that order were more likely than lower doses to cause excessive bleeding. The question would have been whether the data provided sufficient support for a lower once daily dose that would have a sufficiently sustained antithrombotic effect over 24 hours.
- In my judgment, the overall views taken by Professors Hirsh and Wilkins in this regard were the more realistic. The data in Harder and the Kubitza posters gave reasonable grounds for the belief that a once daily dose of 30 mg would be likely to have a sustained effect over 24 hours. The data as a whole offered no reason to believe that such a regimen would cause excessive bleeding. Neither the possibility of excessive bleeding nor insufficient efficacy could be completely ruled out with such a regimen, but as I have discussed, some risk was inevitable.
- Therefore I think that the combined evidence of Professors Hirsh and Wilkins was correct in taking the position that the skilled team would have believed that conducting a phase II trial which included a 30 mg once daily regimen would not have caused an unacceptable level of risk.
- It follows that the skilled team would have found it obvious to conduct a phase II trial which included such a regimen.
- That is sufficient for me to reach a conclusion on inventive step over Harder and the Kubitza posters. I would add, though, that on the evidence presented in this court and so far as one can tell from the Patent specification, Bayer's invention came from a single phase II trial which included a once daily 30 mg regimen. It is to be assumed that this trial was based on Bayer phase I studies. The content of Harder and the Kubitza posters seems to present the assays and results of those phase I studies. If that is right, no reason was advanced to explain why, if Bayer's inclusion of a once daily regimen in phase II was based on the phase I data of Harder and Kubitza, the skilled team would not have done the same thing.
- At one point counsel for Bayer submitted that Bayer must have had more data than is reported in Harder and the Kubitza posters on which to base their design of a phase II study. That may be, but it does not follow that the remaining data added up to anything materially different to what is reported in Harder and the Kubitza posters. Also, it would be odd if there was something materially different and therefore significant to a convincing explanation of the claimed invention, yet Bayer chose not to mention it in this court. Dr Misselwitz was available as a witness, he attended, but he was silent on this topic in his witness statement. In consequence no disclosure was sought from or provided by Bayer in relation to an invention story and Dr Misselwitz was not cross-examined on it.
Perzborn and the Blood Abstracts
- The claimants did not press their case on lack of inventive step over Perzborn and any combination of the Blood Abstracts in the event that I were to find, as I have, that at the priority date the skilled team would have discovered the identity and chemical structure of BAY 59-7939.
Insufficiency
- The pleaded case is that the claims contain no limitation as to dose or types of thromboembolic disorders. Only one once daily dose is disclosed, in Example I, being 30 mg administered once daily for 7-9 days. Accordingly the specification dose not render it plausible that the use of rivaroxaban as claimed will be effective
(a) for the treatment of a thromboembolic disorder in any dose, alternatively any dose other than 30 mg; or
(b) for the treatment of substantially all thromboembolic disorders, or the sub-set identified in claim 2, at substantially all doses, alternatively at any dose other than 30 mg, alternatively at a dose of 30 mg.
The law on insufficiency by reference to plausibility
- The principles to be derived from the concept of plausibility as it has come to be applied in patent law was discussed by Lord Sumption in Generics (UK) Ltd v Warner Lambert Company LLC [2018] UKSC 56, at [37]. Meade J recently reviewed the law in Teva Pharmaceutical Industries Ltd v Grünenthal GmbH [2023] EWHC 1836 (Pat), at [338]-[347].
- The issues of insufficiency raised by the claimants boil down to (a) whether the claims should have been confined to a once daily dose of 30 mg or some other limited range and (b) whether an explanation in the specification of the Patent as to what "thromboembolic disorders" means leads to the result that the claims cover uses of rivaroxaban that would not be effective in relation to some of the conditions listed. These are narrow points which do not require an exploration of the law on plausibility because neither raises the usual issues which arise from an allegation of lack of plausibility.
No limitation as to dose
- Although now qualified by the judgment of the Supreme Court in Actavis UK Ltd v Eli Lilly & Co [2017] UKSC 48 regarding the scope of a claim, Lord Hoffmann's observations on patent construction in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2004] UKHL 46 are still basic principles of law:
"[30]
the author of a document such as a contract or patent specification is using language to make a communication for a practical purpose and that a rule of construction which gives his language a meaning different from the way it would have been understood by the people to whom it was actually addressed is liable to defeat his intentions. It is against that background that one must read the well known passage in the speech of Lord Diplock in Catnic Components Ltd v Hill & Smith Ltd [1982] R.P.C. 183, 243 when he said that the new approach should also be applied to the construction of patent claims:
'A patent specification should be given a purposive construction rather than a purely literal one derived from applying to it the kind of meticulous verbal analysis in which lawyers are too often tempted by their training to indulge.
[32] Construction, whether of a patent or any other document, is of course not directly concerned with what the author meant to say. There is no window into the mind of the patentee or the author of any other document. Construction is objective in the sense that it is concerned with what a reasonable person to whom the utterance was addressed would have understood the author to be using the words to mean.
[33] In the case of a patent specification, the notional addressee is the person skilled in the art. He (or, I say once and for all, she) comes to a reading of the specification with common general knowledge of the art. And he reads the specification on the assumption that its purpose is to both to describe and to demarcate an invention a practical idea
"
- The practical idea of the claims of the Patent is to treat thromboembolic disorders by the administration of rivaroxaban in tablet form no more than once daily. Professors Hirsh and Crowther both found it self-evident that not all doses would be safe and effective. For instance, homeopathic doses would not be effective and therefore not safe either. Very high doses would inevitably not be safe. The skilled person would appreciate that there must be a limited range of doses in the once daily regimen as claimed that will be both safe and effective. The purpose of phase II and III trials would be to find the range. It may vary according to the nature of the thromboembolic disorder which is the subject of the trial. But such trials are routine and within the ordinary scope of the skilled team as appears from the CGK.
- In my view the specification and the CGK of the skilled team would permit the performance of claimed invention across the scope of the claims, the claims being limited with regard to dosage, as would be understood by the skilled team. The claims are not insufficient on this ground.
The over-broad explanation in the specification of thromboembolic disorders
- Paragraph [0024] of the specification begins: "The term 'thromboembolic disorders" includes in particular disorders such as
". There is then a long list of disorders, the clear majority of which are, as was agreed, thromboembolic disorders. It also lists inflammatory diseases, microvascular diseases and Alzheimer's disease. Professors Crowther and Hirsh both said that these three are not thromboembolic disorders and that it was not clear why the specification identified them as such. The claimants did not argue that the understanding of the skilled team would have been any different.
- Sometimes a patent specification will provide a definition of a word or term used in a claim and it will be understood by the skilled person to whom the specification is addressed that the definition is to be strictly applied. But this is not an immutable rule, it will depend on the facts. Where a specification contains an assertion that is plainly wrong according to the CGK of the relevant skilled person, it is liable to be treated as such unless to do so would render the specification or the claims a nonsense.
- In the present case, the specification does not so much make an assertion about the three disorders in issue, rather it puts them into a list where, as the skilled team would appreciate, they do not belong. Their presence in the list would be recognised as an obvious mistake on the part of the patentee. Ignoring the mistake would make sense of the claims. In my view, that is what the skilled team would do and accordingly that is how the claims are to be construed. The claims are not insufficient on this ground either.
Added matter
- The issues of added matter only arise if the claims are found invalid on the ground of insufficiency. I have found that they are not. It follows that Bayer's application to amend the claims falls away.
Conclusion
- The Patent is invalid for lack of inventive step over Harder plus the Kubitza posters.
