Statutory Instruments
2006 No. 2013
Health and safety
The Blood Safety and Quality (Amendment) Regulations 2006
Laid before Parliament
25th July 2006
Coming into force
31st August 2006
The Secretary of State for Health makes these Regulations in exercise of the powers conferred on her by section 2(2) of the European Communities Act 1972(
) and section 56(1) and (2) of the Finance Act 1973(
).
The Secretary of State has been designated(
) for the purposes of section 2(2) of the European Communities Act 1972 in relation to health protection measures regulating the use of material of human origin.
The Treasury has consented to the making of these Regulations as required by section 56(1) of the Finance Act 1973.
Citation, commencement and interpretation
1.—(1) These Regulations may be cited as the Blood Safety and Quality (Amendment) Regulations 2006 and shall come into force on 31st August 2006.
(2) In these Regulations “the principal Regulations” means the Blood Safety and Quality Regulations 2005(
).
Amendment of regulation 1 of the principal Regulations
2. In regulation 1 of the principal Regulations (citation, commencement and interpretation), in paragraph (3)—
(a)
for the definition of “health service hospital”, substitute—
““health service hospital” means a hospital owned or managed by a health service body;
”;
(b)
for the definition of “independent hospital”, substitute—
““independent hospital”—
(a)
in England and Wales, has the same meaning as in section 2 of the Care Standards Act 2000(
)
,
(b)
in Scotland, has the same meaning as in section 2 of the Regulation of Care (Scotland) Act 2001(
)
, and
(c)
in Northern Ireland, has the same meaning as in article 2 of the Health and Personal Social Services (Quality, Improvement and Regulation)(Northern Ireland) Order 2003(
)
”;
(c)
for the definition of “registered person”, substitute—
““registered person” means the person registered as the manager of an independent hospital, a care home or an independent clinic following an application to be registered as such pursuant to—
(a)
section 12(3) of the Care Standards Act 2000,
(b)
section 7(1) of the Regulation of Care (Scotland) Act 2001, or
(c)
article 13(1) of the Health and Personal Social Services (Quality, Improvement and Regulation)(Northern Ireland) Order 2003
”; and
(d)
insert, in the appropriate alphabetical places, the following definitions—
““biomedical research institution” means any body which carries out biomedical research;
“care home”—
(a)
in England and Wales, has the same meaning as in section 3 of the Care Standards Act 2000,
(b)
in Scotland, has the same meaning as in section 2 of the Regulation of Care (Scotland) Act 2001, and
(c)
in Northern Ireland, has the same meaning as in article 2 of the Health and Personal Social Services (Quality, Improvement and Regulation)(Northern Ireland) Order 2003;
“Commission Directive
2005/62/EC
” means Commission Directive
2005/62/EC
of 30th September 2005 implementing Directive
2002/98/EC
of the European Parliament and of the Council as regards Community standards and specifications relating to a quality system for blood establishments(
)
;
“facility” means—
(b)
any other facility or service owned or managed by a health service body,
(d)
an independent clinic,
(f)
a biomedical research institute;
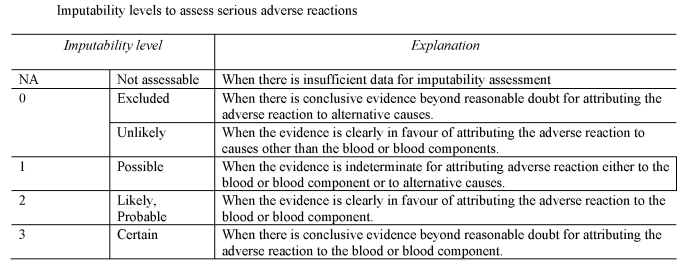
“imputability” means the likelihood that a serious adverse reaction in a recipient can be attributed to the blood or blood component transfused, or that a serious adverse reaction in a donor can be attributed to the donation process;
“independent clinic”—
(a)
in England and Wales, has the same meaning as in section 2 of the Care Standards Act 2000,
(b)
in Scotland, has the same meaning as in section 2 of the Regulation of Care (Scotland) Act 2001, and
(c)
in Northern Ireland, has the same meaning as in article 2 of the Health and Personal Social Services (Quality, Improvement and Regulations)(Northern Ireland) Order 2003;
“issue” means the provision of blood or blood components by a blood establishment or a hospital blood bank for transfusion to a recipient;
“manufacturer” means a person who—
(a)
holds a licence under section 8(2) of the Medicines Act 1968(
)
to manufacture medicinal products;
(b)
holds an authorisation to manufacture an investigational medicinal product granted pursuant to regulation 36 of the Medicines for Human Use (Clinical Trials) Regulations 2004(
)
; or
(c)
falls within the definition of “manufacturer” in paragraph (1) of regulation 2 of the Medical Devices Regulations 2002(
)
;
“person responsible for the management of a facility” means—
(a)
in the case of a hospital, facility or service which is owned or managed by an NHS body, that body,
(b)
in the case of an independent hospital, an independent clinic or a care home, the registered person,
(c)
in the case of a manufacturer or a biomedical research institution, the manufacturer or biomedical research institution;
“person responsible for the management of a reporting establishment” means a blood establishment, the person responsible for the management of a facility or the person responsible for the management of a hospital blood bank;
“recipient” means a person who has been transfused with blood or blood components;
“reporting establishment” means the blood establishment, the hospital blood bank or the facility where the transfusion takes place;
“third country” means any country other than a Member State; and
“traceability” means the ability to trace each individual unit of blood or blood component from the donor to its final destination (whether this is a recipient, a manufacturer of medicinal products or disposal) and from its final destination back to the donor;
”.
Amendment of regulation 3 of the principal Regulations
3.—(1) Regulation 3 of the principal Regulations (requirement for authorisation) is amended as follows.
(2) In paragraph (2)—
(a)
in sub-paragraph (a), at the end, omit “and”;
(b)
in sub-paragraph (b), at the end insert “; and “; and
(c)
after sub-paragraph (b), insert the following sub-paragraph—
“(c)
the import of blood or blood components from a third country
.
”.
(3) In paragraph (3)—
(a)
in sub-paragraph (a), omit “or”;
(b)
in sub-paragraph (b)—
(i)
for “and pursuant”, substitute “or pursuant”; and
(ii)
at the end, insert “; and”; and
(c)
after sub-paragraph (b), insert the following sub-paragraph—
“(c)
the import of blood and blood components from a third country when undertaken by—
(i)
a manufacturer, or
(ii)
a person acting on behalf of and pursuant to a contractual arrangement with a manufacturer,
for the purposes of manufacturing a medicinal product within the meaning of the Medicines act 1968 or the Medical Devices Regulations 2002;
”.
Amendment of regulation 7 of the principal Regulations
4.—(1) In regulation 7 of the principal Regulations (blood establishment requirements), in paragraph (1)—
(a)
in sub-paragraph (b), for “good practice”, substitute “good practice, which complies with the Community standards and requirements set out in the Annex to Commission Directive
2005/62/EC(
);”;
(b)
omit sub-paragraph (e);
(c)
in sub-paragraph (f), at the end, insert “; and”; and
(d)
after sub-paragraph (f), insert the following paragraph—
“(g)
retain, for a period of at least 15 years, a record of any serious adverse events which may affect the quality or safety of blood and blood components.
”.
Amendment of regulation 8 of the principal Regulations
5.—(1) Regulation 8 of the principal Regulations (labelling of blood and blood components and traceability) is amended as follows.
(2) For paragraph (2) substitute—
“(2)
A blood establishment shall maintain, in relation to all blood and blood components collected or prepared by it (including blood and blood components which are imported by it into the European Community)—
(a)
records of the information referred to in paragraph (1) above;
(b)
the records referred to in Part A of Part 6 to the Schedule; and
(c)
such other records as are necessary to ensure full traceability of blood and blood components and identification of each single donation, unit and component.
”.
(3) After paragraph (2), insert the following paragraphs—
“(3)
The records referred to in sub-paragraph (a) shall be maintained—
(a)
in an appropriate and readable storage medium, and
(b)
for a period of not less than 30 years.
(4)
A blood establishment shall ensure that the traceability system in place in the blood establishment enables the tracing of blood and blood components to their location and processing stage.
(5)
A blood establishment shall have in place a system to uniquely identify each donor, each blood unit collected and each blood component prepared, whatever its intended purpose, and the facilities to which a given unit of blood or blood component has been delivered.”.
(6)
A blood establishment shall ensure, when it issues a unit of blood or blood components for transfusion, that the facility to which the unit of blood is issued has in place a procedure to verify that each unit of blood issued has been transfused to the intended recipient or, if not transfused, to verify its subsequent disposition.
”.
Amendment of regulation 9 of the principal Regulations
6.—(1) Regulation 9 of the principal Regulations (hospital blood bank requirements), is amended as follows.
(2) In paragraph (1)—
(a)
in sub-paragraph (b), for “good practice;”, substitute “good practice, which complies with the Community standards and requirements set out the Annex to Commission Directive
2005/62/ECinsofar as these are applicable to hospital blood banks;”;
(b)
for sub-paragraph (e), substitute—
“(e)
maintain in an appropriate and readable storage medium and for a period of not less than 30 years—
(i)
the data set out in Part 6 of the Schedule (insofar as those data are applicable to the activities carried out by the hospital blood bank), and
(ii)
such other data as are needed to ensure full traceability of blood and blood components and the unique identification of each unit of blood and each blood component from the point of receipt of the blood or blood components by the hospital blood bank;
”.
(c)
for sub-paragraph (f), substitute the following sub-paragraph—
“(f)
retain, for a period of not less than 30 years, a record of any serious events which may affect the quality or safety of blood or blood components;
”;
(d)
in paragraph (g), at the end, omit “and”;
(e)
in paragraph (h), at the end, insert “; and”; and
(f)
after paragraph (h), insert the following paragraphs—
“(i)
ensure that the traceability system in place in the hospital blood bank enables the tracing of blood components to their final destination; and
(j)
where it delivers blood or blood components for transfusion at another facility, have in place a system to uniquely identify the facility to which a given unit of blood or blood component has been delivered.
”.
(3) After paragraph (1), insert the following paragraph—
“(2)
A person responsible for management of a hospital blood bank shall ensure that when a hospital blood bank issues a unit of blood for transfusion, that it has in place a procedure to verify that each unit of blood issued has been transfused to the intended recipient, or if not transfused, to verify its subsequent disposition.
”.
Insertion of regulations 12A and 12B
7. After regulation 12 (objections to suspensions, revocations atc.) insert the following regulations—
“Requirement that facilities retain certain data
12A.—
(1)
A person responsible for management of a facility shall ensure that the facility—
(a)
retains the data set out in Section B of Part 6 of the Schedule, in an appropriate and readable storage medium, for a period of at least 30 years; and
(b)
has in place a system in place to record each unit of blood or blood component received, whether or not locally used, and the final destination of that received unit whether transfused, used in the manufacture of medicinal products, discarded or returned to the blood establishment or hospital blood bank.
Requirement to report serious adverse reactions and events
12B.—
(1)
A person responsible for management of a reporting establishment shall ensure that the reporting establishment—
(a)
has in place procedures to retain the record of transfusions for a period of at least 30 years
;
(b)
notifies blood establishments without delay of any serious adverse reactions observed in recipients during or after transfusion which may be attributable to the quality or safety of blood or blood components; and
(c)
notifies the Secretary of State as soon as is known all relevant information about suspected serious adverse reactions using the notification formats set out in Section A and Section C of Part 7 of the Schedule.
(2)
A person responsible for management of a reporting establishment shall ensure that the reporting establishment
—
(a)
notifies the Secretary of State of all relevant information about serious adverse reactions of imputability level 2 and 3 as referred to in Section B of Part 7 of the Schedule, which may be attributable to the quality and safety of blood or blood components;
(b)
notifies the Secretary of State, as soon as is known, of any case of transmission of infectious agents by blood or blood components;
(c)
as part of the notification referred to in paragraph (a), describes the actions taken with respect to other implicated blood or blood components that have been distributed for transfusion or for plasma fractionation;
(d)
as soon as is reasonably practicable after each suspected serious adverse reaction, evaluates that reaction according to the imputability levels set out in Section B of Part 7 of the Schedule;
(e)
completes the serious adverse reaction notification, upon conclusion of the investigation, using the format set out in Section C of Part 7 to the Schedule; and
(f)
submits a complete report to the Secretary of State on serious adverse reactions in any calendar year by no later than 1st April in the following calendar year, using the format set out in Section D of Part 7 to the Schedule.
(3)
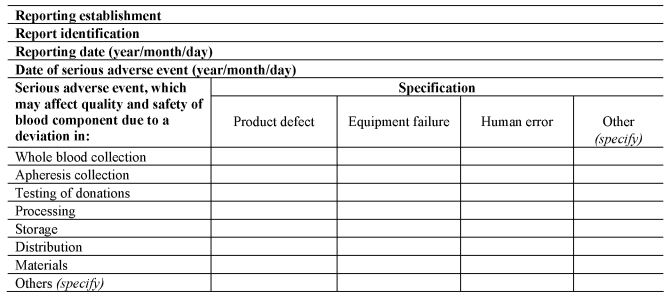
A person responsible for management of a reporting establishment shall ensure that the reporting establishment notifies the Secretary of State as soon as is known, using the notification formats set out in Section A of Part 8 of the Schedule, of all relevant information about serious adverse events which may put in danger donors or recipients other than those directly involved in the event concerned.
(4)
A person responsible for management of a reporting establishment shall ensure that the reporting establishment—
(a)
as soon as is reasonably practicable after each serious adverse event, evaluates that serious adverse event to identify preventable causes within the process;
(b)
upon completion of the investigation, completes the serious adverse event notification, using the format set out in Section B of Part 8 of the Schedule; and
(c)
submits a complete report to the Secretary of State on serious adverse reactions in any calendar year by no later than 1st April in the following calendar year, using the format set out in Section D of Part 7 to the Schedule.
(5)
Provided that either the condition set out in paragraph (6)(a), or the conditions set out in paragraph (6)(b) and (c) are satisfied, a facility may make arrangements with a hospital blood bank for the hospital blood bank to submit to the Secretary of State or the blood establishment the reports required by paragraphs (1)(b) and (c), (2)(a),(b),(e) and (f) and (3)(b) and(c) on the facility’s behalf.
(6)
The conditions referred to in paragraph (5) are that—
(a)
the person responsible for management of the hospital blood bank is the same person as the person responsible for management of the facility with which the arrangement is made; or
(b)
the arrangements referred to in paragraph (5) must be—
(i)
evidenced by a written agreement, and
(ii)
made with the person responsible for management of the hospital blood bank who supplied the blood or blood components to the facility for transfusion; and
(c)
the facility must supply the information necessary to enable the hospital blood bank to make the reports within the timescale specified by this regulation in relation to that report .
”.
Amendment of regulation 13 of the principal Regulations
8. For regulation 13 of the principal Regulations (import of blood and blood components into the United Kingdom) substitute—
“13.
Any person who imports blood or blood components into the United kingdom from a third country must ensure that each unit of blood and each blood components which he imports—
(a)
has been prepared in accordance with standards equivalent to the Community standards and requirements set out in the Annex to Commission Directive
2005/62/EC
; and
(b)
meets standards of quality and safety equivalent to those laid down in Part 5 of the Schedule.
”.
Amendment of regulation 15 of the principal Regulations
9. In regulation 15 of the principal Regulations (inspections, etc.)—
(a)
in paragraph (5), after “blood banks”, in each place where those words appear, insert “and facilities”;
(b)
in paragraph (6), after “blood bank”, in each place where those words appear insert “or a facility”; and
(c)
in paragraph (7), after “hospital blood bank”, insert “or a facility”.
Amendment of regulation 16 of the principal Regulations
10. In regulation 16 of the principal Regulations (records to be kept by the Secretary of State), in paragraph (2)—
(a)
after “management of hospital blood banks”, insert “and facilities”;
(b)
after “otherwise”, omit “or”; and
(c)
after “relating to hospital blood banks”, insert “or facilities”.
Insertion of regulation 16A
11. After regulation 16 (records to be kept by the Secretary of State), insert the following regulation—
“Requirement that the Secretary of State communicate certain information to other competent authorities
16A.
The Secretary of State shall communicate to the competent authorities of other Member States such information as is appropriate with regard to serious adverse reactions and events in order to guarantee that blood or blood components known or suspected to be defective are withdrawn from use and discarded.
”.
Amendment of regulation 17 of the principal Regulations
12. In regulation 17 of the principal Regulations (powers of entry, etc.), in paragraph (1)(a)—
(a)
in head (ii)—
(i)
for “and” in the first place where that term appears, substitute “or”, and
(ii)
at the end, omit “and”;
(b)
in head (iii), at the end, insert “and”; and
(c)
after head (iii), insert the following head—
“(iv)
any premises where transfusion of blood or blood components takes place, or which are owned or managed by a person responsible for management of a facility to which blood or blood components have been delivered.
”.
Amendment of regulation 18 of the principal Regulations
13. In regulation 18 of the principal Regulations (criminal offences), in paragraph (1), after sub-paragraph (e), insert the following sub-paragraphs—
“(f)
regulation 12A, and
(g)
regulation 12B,
”.
Amendment of regulation 22 of the principal Regulations
14. In regulation 22 of the principal Regulations (fees)(
)—
(a)
in paragraph (3B)—
(i)
for “paragraph (3D)” substitute “paragraphs (3D) and (3E)”;
(ii)
after “hospital blood banks” insert “or facilities”; and
(iii)
after “hospital blood bank”, insert “or facility”;
(b)
after paragraph (3D), insert the following paragraph—
“(3E)
No fee shall be payable under paragraph (3B) by a person responsible for the management of a facility where the facility makes arrangements with a hospital blood bank, pursuant to regulation 12B(5) that the hospital blood bank will report serious adverse reactions and events to the Secretary of State on behalf of the facility.
”,
(c)
in paragraph (4)—
(i)
after “hospital blood bank”, in the first place where those words appear, insert “or a facility”; and
(ii)
after “hospital blood bank”, in the second place where those words appear, insert “or the facility”; and
(d)
in paragraph (5), after “hospital blood banks”, insert “or facilities”.
Amendment of the Schedule to the principal Regulations
15. In the Schedule to the principal Regulations, after Part 5 (quality and safety requirements for blood and blood components) insert the following Parts—
“PART
6
RECORD OF DATA ON TRACEABILITY
A. BY BLOOD ESTABLISHMENTS
1.
Blood establishment identification
2.
Blood donor identification
3.
Blood unit identification
4.
Individual blood component identification
5.
Date of collection (year/month/day)
6.
Facilities to which blood units or blood components are distributed, or subsequent disposition.
B. BY FACILITIES
1.
Blood component supplier identification
2.
Issued blood component identification
3.
Transfused recipient identification
4.
For blood units not transfused, confirmation of subsequent disposition
5.
Date of transfusion or disposition (year/month/day)
6.
Lot number of the component, if relevant.
PART 7
NOTIFICATION OF SERIOUS ADVERSE REACTIONS
SECTION A
Rapid notification format for suspected serious adverse reactions
SECTION B
Serious adverse reactions – imputability levels
SECTION C
Confirmation format for serious adverse reactions
SECTION D
Annual notification format for serious adverse reactions
PART 8
NOTIFICATION OF SERIOUS ADVERSE EVENTS
SECTION A
Rapid Notification Format for Serious Adverse Events
SECTION B
Confirmation Format for Serious Adverse Events
SECTION C
Annual Notification Format for Serious Adverse Events
Signed by authority of the Secretary of State for Health
Caroline Flint
Minister of State
Department for Health
18th July 2006
We consent,
Alan Campbell
Dave Watts
Two of the Lords Commissioners of Her Majesty’s Treasury
24th July 2006
EXPLANATORY NOTE
These Regulations further amend the Blood Safety and Quality Regulations 2005 (“the principal Regulations”). The principal Regulations implement Directive
2002/98/ECof the European Parliament and Council setting out the standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components (“the Directive”)(
) They also implement Commission Directive
2004/33/EC(
), which contains certain technical requirements relating to blood and blood components.
These Regulations further amend the principal Regulations to implement Commission Directive
2005/61/EC(
) and Commission Directive
2005/62/EC(
) which contain further technical requirements with regard to blood and blood components.
Regulation 2 amends regulation 1 of the principal Regulations to insert further definitions and to amend some of the definitions in the principal Regulations.
Regulation 3 amends regulation 3 of the principal Regulations to provide that the import of blood or blood components from a country outside the European Community may only be undertaken by a blood establishment or by a licensed manufacturer of medicines or a manufacturer of medical devices.
Regulation 4 amends regulation 7 of the principal Regulations to provide that the quality system maintained by blood establishments must comply with the requirements of Commission Directive
2005/62/ECand to require that blood establishments retain records of serious adverse events.
Regulation 5 amends regulation 8 of the principal Regulations to impose further requirements on blood establishments with regard to traceability (the tracing of individual blood donations from donor to recipient and vice versa) in accordance with the requirements of Commission Directive
2005/61/EC.
Regulation 6 amends regulation 9 of the principal Regulations to impose further requirements on hospital blood banks with regard to traceability in accordance with the requirements of Commission Directive
2005/61/EC, and to provide that the quality system maintained by hospital blood banks must comply with the requirements of Commission Directive
2005/62/EC.
Regulation 7 inserts new regulations 12A and 12B into the principal Regulations. Regulation 12A requires that facilities which receive blood (i.e care homes, independent clinics, hospitals and other NHS facilities and services, manufacturers of medicines and medical devices and biomedical research institutes) keep certain records. Regulation 12B requires that facilities which undertake blood transfusions report adverse events and reactions to the blood establishment from which the blood involved in the adverse incident was received, and to the Secretary of State.
Regulation 8 amends regulation 13 of the principal Regulations to provide that any person who imports blood and blood components imported into the European Community must ensure that that blood and those blood components have been prepared to equivalent standards to those set out in Commission Directive
2005/62/EC.
Regulations 9 and 10 extend the Secretary of State’s obligations regarding inspection and record keeping, respectively, to include facilities. Regulation 11 imposes an obligation on the Secretary of State to notify details of serious adverse reactions and events to the competent authorities of other Member States in appropriate cases.
Regulations 12 to 14 amend regulations 17, 18 and 22 of the principal Regulations to provide, respectively, that the Secretary of State shall have power of entry into a facility, that breach of the obligations placed on facilities by regulations 12A and 12B shall be a criminal offence and that a fee shall be payable by a facility in respect of heamovigilance (except where a facility has made arrangements with a hospital blood bank that the blood bank will report adverse incidents on the facility’s behalf) and for an inspection under these Regulations. Haemovigilance is the monitoring of serious adverse incidents by the Secretary of State in order to ensure that potentially contaminated products are removed from the distribution chain.
Regulation 15 provides for the Schedule to the principal Regulations to be amended to include certain technical requirements relating to haemovigilance.
A full Regulatory Impact Assessment of the effect that this instrument will have on the costs of business, and a Transposition Note in relation to the implementation of Directives
2005/61/ECand
2005/62/EChave been placed in the libraries of both Houses of Parliament and copies may be obtained from Department of Health, Area 530, Wellington House, 133-155 Waterloo Road London SE1 8UG.