Contents |
|
| |
|
| Topic |
Paragraphs |
| Introduction |
1 |
| The witnesses |
2-13 |
| The Claimants' technical experts |
3-6 |
| The Defendants' technical experts |
7-10 |
| Experts in Japanese |
11 -13 |
| Technical background |
14-62 |
| The human genome |
15-23 |
| Genetic polymorphism |
24-25 |
| Genetic disorders |
26-27 |
| Mendelian disorders |
28 |
| X chromosome-linked recessive disorders |
29-30 |
| Blood |
31-33 |
| Cell-free DNA |
34 |
| Mechanisms of cell death |
35-36 |
| Pre-natal development |
37-39 |
| The placenta |
40-44 |
| Cytogenetic analysis of foetal cells |
45-47 |
| Chorionic Villus Sampling |
48-49 |
| Amniocentesis |
50-51 |
| Foetal blood and tissue sampling |
52-53 |
| Foetal cell-free DNA |
54-55 |
| Polymerase chain reaction |
56-59 |
| Quantitative PCR |
60 |
| Gel electrophoresis |
61 |
| Centrifugation |
62 |
| The Patent |
63-73 |
| The claims |
74-75 |
| The skilled team or person |
76-77 |
| Common general knowledge |
78-107 |
| Cell-free DNA |
79-80 |
| Extraction of cell-free DNA |
81-82 |
| Efforts to improve detection of foetal cell-free DNA |
83-86 |
| Quantitative PCR |
87-107 |
| Fluorescent reporters |
90-91 |
| Amplification efficiency |
92-95 |
| Reference genes |
96 |
| Sensitivity and specificity |
97 |
| Statistics |
98 |
| Relative vs absolute quantification |
99-101 |
| Standard curves |
102-103 |
| Copy number |
104-105 |
| Resolution |
106-107 |
| Construction |
108-115 |
| Extracellular DNA |
109-111 |
| Two-fold enrichment |
112-113 |
| Size separation |
114-115 |
| Ikeda |
116 |
| The translation issue |
117-129 |
| What does Ikeda disclose? |
130-190 |
| The skilled person's approach to Ikeda |
133-134 |
| Hindsight |
135-136 |
| The title |
138 |
| The objective |
139-143 |
| The conclusion |
144-147 |
| The method |
148-151 |
| The results |
152-160 |
| Frequency of SRY relative to beta-globin |
161-190 |
| Single reference gene |
164-166 |
| Confidence in the data |
167-168 |
| The design of the experiment and foetal fractions |
179-190 |
| Obviousness over Ikeda |
191-192 |
| Insufficiency |
193-241 |
| The law |
194-198 |
| The Defendants' case in outline |
199-201 |
| Chan 2004 |
202-213 |
| Li 2004 |
214-232 |
| Cheng 2015 |
233-236 |
| Electropherograms |
237 -238 |
| Conclusions |
239 -241 |
| Discovery as such |
242-245 |
| Infringement |
246 |
| Summary of principal conclusions |
247 |
Introduction
1. The Second Claimant is the proprietor of European Patent (UK) No. 1 524 321 entitled “Non-invasive detection of fetal genetic traits” (“the Patent”), which has a filing date of 16 October 2003. The First Claimant has been an exclusive licensee of the Patent since at least 11 January 2019. The Third Defendant (“Ariosa”), which is a member of the Roche group of companies, has developed a non-invasive prenatal test (“NIPT”) called the Harmony Test. The First Defendant (“TDL”) offers the Harmony Test to patients, and uses it. The Claimants allege that TDL has thereby infringed the Patent. Ariosa admits that it is jointly liable for any infringement by TDL. The Defendants dispute that the Harmony Test infringes the Patent and counterclaim for revocation of the Patent on the grounds of obviousness over Ikeda et al , “Frequency at which foetal DNA is present in maternal plasma: Difference by fragment length”, J Japan Soc of Obs & Gyn , 55(2), P-910 (2003) (“Ikeda”) and insufficiency. A number of other issues which had been raised by the Defendants were not pursued in closing submissions.
The witnesses
2. Both sides called two technical experts, but the two pairs of experts were not matching pairs. In addition, each side called an expert in Japanese to address an issue concerning the translation of Ikeda.
The Claimants’ technical experts
3. Since 2015 Professor William Allen Hogge has been a Professor in the Department of Obstetrics and Gynaecology at Virginia Commonwealth University. He obtained a BA in Biology from the University of Virginia in 1970 and an MD from the same institution in 1973. After a residency in Obstetrics and Gynaecology at University of Virginia Hospital and a spell in private practice, he held a series of Assistant and Associate Professor positions in departments of obstetrics and gynaecology from 1982 to 1992. From 1992 to 2014 he was Professor in the Department of Obstetrics and Gynaecology at the University of Pittsburgh (“UP”) School of Medicine. From 1992 he was also an Associate Professor of Human Genetics and Director of Reproductive Genetics at UP and Medical Director of the Department of Genetics at the Magee-Women’s Hospital in Pittsburgh (“MWH”). From 1997 to 2013 he was Director of the Pregnancy Screening Laboratory at MWH. From 2003 to 2014 he was the Director of the Center for Medical Genetics and Genomics for UP and the Chair of the Department of Obstetrics and Reproductive Sciences at the UP School of Medicine. From 2010 to 2014 he was also a Professor in the Department of Pathology at the UP School of Medicine. His clinical work has involved carrying out prenatal diagnosis procedures, such as amniocentesis and CVS, and he has been involved in research relating to methods of non-invasive prenatal diagnosis. He is the author or co-author of 19 books and over 80 scientific publications relating to prenatal diagnosis.
4. Counsel for the Defendants accepted that Prof Hogge had given evidence in a measured manner, and made no criticism of him. Counsel submitted that Prof Hogge’s relevant personal experience was more limited than that of the Defendants’ experts, but accepted that Prof Hogge was aware of the state of, and thinking in, the cell-free DNA field at the filing date.
5. Since 2013 Professor Michael Lovett has been Professor of Systems Biology at the National Heart and Lung Institute at Imperial College, London. He obtained a BSc in Molecular Biology from the University of Edinburgh in 1977 and a PhD in Biochemistry from Imperial in 1981. From 1982 to 1987 he was first a Postdoctoral Fellow and then Assistant Professor in Genetics at the University of California, San Francisco. From 1987 to 1992 he worked for Genelabs Inc. From 1992 to 1999 he was an Associate Professor of Biochemistry at the University of Texas Southwestern Medical Centre. From 1999 to 2013 he was Professor of Genetics and Human Genetics Division Head at the Washington University School of Medicine in St Louis, which was one of the world’s five primary Genome centres. In 2003 he was conducting work on methods of direct selection of defined regions of genomic material, enabling the targeted capture and sequencing of genes of interest and the discovery of mutations in Mendelian disorders. Professor Lovett is the author or co-author of over 100 scientific publications relating to mammalian molecular genetics and genomics.
6. Counsel for the Defendants did not criticise Prof Lovett as a witness, but submitted that it was important to appreciate that Prof Lovett’s background was in gene expression and that (unlike Dr Daniels) Prof Lovett was not at the filing date, and never had been, in the cell-free DNA field. I accept that this is a relevant factor for me to take into account, but on the other hand it was not suggested that Prof Lovett was not in a position to assist the Court as to how the person skilled in the art would understand Ikeda.
The Defendants’ technical experts
7. Prior to his retirement in October 2015, Dr Geoff Daniels was the Head of Diagnostics at the International Blood Group Reference Laboratory (“IBGRL”) in Bristol and Senior Research Fellow at the Bristol Institute for Transfusion Sciences (“BITS”). He obtained a BSc in Zoology from the University of Aberdeen in 1972 and a PhD from the University of London in 1980. From 1972 to 1973 he worked at the South London Transfusion Centre. From 1973 to 1995 he worked at the Medical Research Council Blood Group Unit. From 1995 to 2015 he worked at IBGRL, becoming Head of Molecular Diagnostics in 2001 and Head of Diagnostics in 2012, and at BITS. From 1988 to 2015 he also had teaching responsibilities at the University of Bristol. From 2001 around half of his time was spent managing a blood group phenotyping service (about half of which involved non-invasive prenatal testing of blood group antigens using maternal plasma) and half directing a small research team and teaching. In 2001 he collaborated on the establishment of a service for non-invasive prenatal testing for RhD on foetal cell-free DNA in maternal plasma using quantitative PCR based on Lo 1998 (as to which, see below). In 2003 most of the samples received by IBGRL for pre-natal testing were for RhD, but some were for testing for Y-chromosome markers. Dr Daniels is the author of one book and co-author of another book both on blood groups and published over 200 scientific papers during his career.
8. Counsel for the Claimants accepted that Dr Daniels had given his oral evidence very fairly. Counsel submitted that Dr Daniels’ evidence on Ikeda was tainted by hindsight and by personal knowledge he had had at the filing date, points which it is to convenient to consider in context, but made it clear that this was not a criticism of Dr Daniels personally.
9. Since 2008 Professor Alain Thierry has been the Director of Research at INSERM at the Institut de Recherche en Cancérologie de Montpellier. He obtained a BSc in Biological Sciences and Technology from Université de Clermont in 1981, an MSc from Université de Clermont-Ferrand II in 1983 and a PhD from Université Montpellier 2 in 1986. From 1986 to 1992 he held post-doctoral positions at Université de Clermont-Ferrand II and the Lombardi Cancer Center, Georgetown University Medical Center, and from 1992 to 1996 he was Adjunct Assistant Professor at the latter institution. From 1997 to 2001 he was Scientific Director in the Department of Gene Therapy and Delivery at Biovector Therapeutics. From 2001 to 2007 he was Associate Professor at Université Montpellier 2, where he worked on gene delivery and gene therapy and the biophysics of DNA complexes. Since 2005, his research has focused on the size, structure and origins of circulating cell-free DNA, and on the application of methods to detect circulating cell-free DNA, especially in the field of oncology. He is the author of over 20 peer-reviewed publications relating to circulating cell-free DNA.
10. The Defendants mainly relied upon the evidence of Prof Thierry in support of their insufficiency case. Counsel for t he Claimants submitted that Prof Thierry was a man on a mission. Counsel accepted that Prof Thierry’s zeal was entirely genuine, but submitted that the question for the Court was whether his opinions were supported by the available data. I agree with this.
Experts in Japanese
11. The Claimants’ expert was Professor Peter Kornicki. Prof Kornicki is a native English speaker who has been Emeritus Professor of Japanese Studies at the University of Cambridge since 2014. He obtained a BA in Japanese and Korean from the University of Oxford in 1972, followed by an MSc in 1975 and a DPhil in Japanese literature in 1979. From 1978 to 1982 he taught at the University of Tasmania and from 1982 to 1985 at Kyoto University. Between 1985 and 2014 he was successively a lecturer, reader in Japanese History and Bibliography, Professor of Japanese Studies and Professor of East Asian Studies at Cambridge. Among other honours, he has been elected a Fellow of the British Academy, awarded the degree of Doctor of Letters by the University of Oxford and the Order of the Rising Sun with Gold Rays and Neck Ribbon by the Emperor of Japan. He has published a number of books and many articles on Japanese culture, and in particular Japanese books.
12. The Defendants’ expert was Professor Yoshifumi Itoh. Prof Itoh is a native Japanese speaker who has been a Senior Lecturer at the Kennedy Institute of Rheumatology since 2001, initially at Imperial College, London and since 2011 at the University of Oxford. Prof Itoh was educated in Japan, and studied English as a second language. He obtained a BSc in Pharmacy from Tokyo College of Pharmacy in 1989, an MSc in Clinical Pharmacy (Biochemistry) from the same institution in 1991 and a PhD from the same institution in 1996. From 1991 to 1997 he worked first as a Research Assistant and then as a Postdoctoral Fellow at the University of Kansas Medical Centre. From 1997 to 2001 he was Assistant Professor in the Department of Cancer Research, Institute of Medical Science, University of Tokyo. During the course of his career, Prof Itoh has worked extensively in both Japanese and English, including both writing and reviewing many scientific articles in both languages.
13. It is common ground that both Prof Kornicki and Prof Itoh did their best to assist the Court. Counsel for the Claimants submitted that Prof Kornicki was better placed to do so because Prof Kornicki’s expertise is (among other things) as a Japanese linguist, whereas Prof Itoh is not a linguist. Moreover, Prof Kornicki is a native English speaker, and it is generally accepted that translators should translate into their mother tongue. Counsel for the Defendants submitted that Prof Itoh was better placed to assist the Court since he is a native Japanese speaker familiar with how scientific abstracts are written in both Japanese and English. In my view it was of assistance to have evidence from both perspectives.
Technical background
14. The following account of the technical background is mainly based on the primer helpfully agreed by the parties.
The human genome
15. The human genome represents the complete set of inherited instructions encoded in deoxyribonucleic acid (DNA) in a human cell. The human haploid genome (i.e. one set of chromosomes) consists of approximately 3 billion base pairs of DNA. In typical diploid individuals (i.e. individuals with a balanced pairing of chromosomes) the genome is organised into a total of 46 chromosomes.
16. Chromosomes consist of DNA and protein complexes. Chromosomal DNA consists of two complementary strands which are coiled to form a double helix. The DNA double helix is tightly wound around histone proteins to form complexes called nucleosomes. Nucleosomes fold up to form chromatin fibres which coil to form the chromatid of a chromosome. The structural organisation of a chromosome is depicted in Figure 1 below. During processes such as transcription and replication the chromatin fibres are opened up and/or the histones are removed transiently to permit transcription or replication to proceed.

17. The strands of DNA are made up of nucleotides, which are composed of a phosphorylated sugar (deoxyribose) backbone, each sugar unit being attached to a nitrogenous base. There are four different nitrogenous bases in DNA: adenine (A), cytosine (C), guanine (G), or thymine (T). The two complementary strands are held together by (amongst other interactions) base-pairing with formation of hydrogen bonds between the bases, where the general rule is that cytosine (C) only pairs with guanine (G), and adenine (A) only pairs with thymine (T). Cytosine (C) pairs with guanine (G) by three hydrogen bonds, whereas adenine (A) pairs with thymine (T) by two hydrogen bonds.

18. Chromosomal DNA is replicated in the human cell nucleus. As explained above, for this to occur the DNA-protein complexes must be disassembled and the DNA strands temporarily separated in the region of DNA being replicated. DNA helicases catalyse enzyme-dependent separation of the complementary strands from the site of previously bound initiator proteins, allowing DNA polymerase enzyme activity to synthesise two new strands using free deoxynucleoside triphosphates (dNTPs). With the incorporation of each deoxynucleotide into the growing DNA strand, a pyrophosphate (two phosphate groups linked together) is released. Each strand of the original DNA molecule acts as a template for the production of a complementary strand in order to form two copies of the original DNA molecule. A schematic representation of DNA replication is shown in Figure 3 below.

19. Genes are functional units of DNA in the genome that code for particular proteins and non-coding RNAs. Protein-coding genes may be “transcribed” to produce messenger ribonucleic acid (mRNA), which in turn may be “translated” to produce the protein encoded by the gene.
20. The 46 chromosomes in a normal human somatic cell (cells other than gametes (spermatozoa and ova (egg cells)), germ cells (cells which give rise to gametes), gametocytes and undifferentiated stem cells) are made up of 22 pairs of homologous autosomes (non-sex chromosomes) and two allosomes (sex chromosomes – XX for typical females and XY for typical males). One set of 23 chromosomes is maternally-inherited and one set of 23 chromosomes is paternally-inherited. The autosomes (and the X chromosomes in the case of typical XX females) are said to be diploid (or paired).
21. Each chromosome carries a set of genes. In normal somatic cells, autosomal genes are present in pairs, one gene being maternally-inherited and one paternally-inherited. Each gene is encoded at a specific site or “locus” on a chromosome. Different versions of a gene (for example, caused by variants such as single or multiple base changes) may be referred to as “alleles”. Where an individual has two copies of the same allele (i.e. the same allele at a particular locus on each chromosome within one pair), they are said to be “homozygous” with respect to that allele; where the alleles are different (i.e. there are different alleles at a particular locus on each chromosome within one pair), the individual is said to be “heterozygous” with respect to that allele. An individual is said to be hemizygous when they have only one allele, rather than the typical two (which may arise, for example, where only one chromosome of a pair is present, where one copy of an allele has been deleted or, in normal male somatic cells, in respect of certain genes contained on the allosomes).
22. Normal male somatic cells contain a maternally-inherited X chromosome and a paternally-inherited Y chromosome, whilst normal female somatic cells contain two X chromosomes, one maternally-inherited and one paternally-inherited. Normal gametes (spermatozoa and ova) are haploid, which means they contain only one copy of each chromosome. Normal ova carry one X allosome. Approximately 50% of spermatozoa contain one X allosome and 50% contain one Y allosome.
23. Typically, the mother’s allosome genotype is XX, so she passes an X chromosome to her offspring and the father’s allosome genotype is XY, so he passes either an X chromosome to his offspring (resulting in a female child with an XX genotype), or a Y chromosome (resulting in a male child with an XY genotype). However, there are a number of known aberrant genetic conditions involving the allosomes, including aneuploidies.
Genetic polymorphism
24. The existence of different variants at a locus is referred to as genetic polymorphism. The presence of a particular variant at a polymorphic locus can act as a marker. Whilst some variants give rise to distinct biological phenotypes (i.e. observable traits or characteristics of individuals such as eye colour or blood group), others have no or unknown phenotypic effect.
25. Different alleles may arise via single base changes or multiple base changes. Single base changes that occur with at least a certain prevalence in a population, are now known as single nucleotide polymorphisms or SNPs. Genotyping means identifying differences between the DNA of an individual and the general population or other specific individuals.
Genetic disorders
26. Genetic disorders may be caused by pathogenic changes in the genome. Genetic disorders include Mendelian (or single-gene) disorders, where a single gene is altered, or chromosomal disorders, where an entire chromosome, or a large segment of it, is deleted, duplicated, translocated or otherwise altered.
27. Genetic disorders may be inherited, or they may arise for the first time in the egg, spermatazoa or fertilised egg (termed “de novo”), or they may arise during a person's lifetime (termed “acquired”).
Mendelian disorders
28. Mendelian disorders (also known as single gene disorders) are genetic disorders caused by a mutation in a single gene. Autosomal recessive single gene disorders occur in individuals with mutations in both alleles of a gene (i.e. individuals who are homozygous for the mutation). An individual with a single copy of the mutant allele (i.e. individuals who are heterozygous for the mutation) is referred to as a “carrier” of the disorder. Examples of autosomal recessive single gene disorders include cystic fibrosis and haemoglobinopathies. Autosomal dominant single gene disorders require only a single copy of the mutant allele and therefore occur in both heterozygous and homozygous individuals. Examples of autosomal dominant single gene disorders include Huntington’s chorea and Marfan syndrome.
X chromosome-linked recessive disorders
29. X chromosome-linked recessive disorders are genetic disorders which occur due to a mutation on the X chromosome. X chromosome-linked recessive disorders occur in males who are hemizygous for the gene mutation (since men possess a single X chromosome) or females who are homozygous for the gene mutation (i.e. both copies of their X chromosomes possess the mutation). However, it is typically males who are affected by X chromosome-linked recessive disorders, in part due to the greater likelihood of inheriting a single copy of the mutation than of inheriting two copies. Examples of X chromosome-linked recessive disorders include haemophilia, Duchenne muscular dystrophy and fragile X syndrome.
30. The presence of a variation in the number of chromosomes from the usual complement (i.e. 46 chromosomes) is referred to as aneuploidy. The absence of a single chromosome from a usual pair is referred to as monosomy, and the presence of an additional copy of a single chromosome to a usual pair is referred to as trisomy. The additional chromosome may be stand-alone or may be bound to another chromosome. The additional chromosome may be paternally- or maternally-inherited. The most common form of aneuploidy (trisomy of chromosome 21) results in Down’s syndrome.
Blood
31. Whole blood contains blood cells and a liquid portion. Blood cells include oxygen-carrying erythrocytes (red blood cells), immune cells called leukocytes or white blood cells, and thrombocytes (platelets), which regulate blood clotting. Mature erythrocytes are anucleated, i.e. they do not contain a nucleus or chromosomal DNA. Immature nucleated red blood cells are also found in the blood of foetuses, but are rapidly cleared from the bloodstream after birth.
32. The liquid portion of blood can be obtained either as plasma, when the blood sample is treated with an anticoagulant, or serum, when blood is allowed to clot. Plasma is a straw-yellow fluid which contains water, blood plasma proteins (including clotting factors), cell-free DNA and minerals, as well as dissolved nutrients (such as glucose, amino acids, and fatty acids) and waste products (such as urea and lactic acid).
33. Whole blood may be separated by centrifugation (which is described in more detail below), which in general terms and depending on the experimental conditions results in the formation of three layers: (i) the upper plasma layer; (ii) the intermediate buffy coat layer, which contains leukocytes and thrombocytes; and (iii) the lower layer, which contains erythrocytes. Chromosomal DNA may be extracted from the buffy coat layer, which contains nucleated cells, and the plasma layer, which contains cell-free DNA.

Cell-free DNA
34. Cell-free fragments of nucleic acids, including DNA, are present in the blood plasma and serum of human beings. Cell-free nucleic acids can originate from various sources, including cell death.
Mechanisms of cell death
35. Necrosis is a mechanism of cell death that arises due to infection, toxins or trauma. Necrosis involves the loss of integrity of the cell membrane and the uncontrolled release of the products of cell death.
36. Apoptosis (or programmed cell death) is a highly regulated process of cell death that can be initiated through a number of signalling pathways. Apoptosis is characterised by characteristic cell morphology, including: (i) blebbing (the formation of protrusions in the cell membrane known as “blebs”); (ii) cell shrinkage; (iii) nuclear condensation (pyknosis) and fragmentation (karyorrhexis); (iv) chromatin condensation; and (v) chromosomal DNA fragmentation.
Pre-natal development
37. Following fertilisation of an ovum (egg) by a spermatozoon in the Fallopian tubes, the resulting single cell zygote travels down the Fallopian tube and divides to form a blastocyst. Approximately five days after fertilisation, the blastocyst, which consists of trophoblast cells and embryonic cells, reaches the uterus and becomes embedded in the endometrium (lining) of the uterus.
38. The trophoblast cells, which surround the embryonic cells, proliferate and embed further into the uterine lining, eventually forming the placenta (described below). The blastocyst becomes fully implanted approximately 7-12 days after fertilisation.
39. At the beginning of the second week, the formation of the fluid-filled amniotic sac starts. Foetal development takes place in the amniotic sac, which cushions the foetus and provides buoyancy.
The placenta
40. The placenta is a composite structure made up of maternal tissues as well as those derived from the foetus. Foetal blood vessels extend to the placenta via the umbilical cord and branch into many chorionic villi, providing a large surface area for the exchange of materials between foetal and maternal blood across a layer of tissue called the placental membrane. A variety of materials, including nutrients and oxygen, are exchanged between the maternal circulatory system and the foetus via chorionic villi in the placenta and the umbilical cord. Other materials passing from the foetus or placenta into the maternal blood circulation include foetal blood cells, proteins and hormones which form the basis of the Rh disease test. Likewise, waste materials are removed from the foetus to the maternal circulation.

Rhesus haemolytic disease
41. Rhesus (Rh) factor (also known as the Rh D antigen) is a protein found on the surface of red blood cells in so called Rh positive individuals. Rh negative individuals lack this protein. Lack of this protein in Rh negative individuals is caused by a deletion or mutations of the gene (RhD) that encodes it in both copies of chromosome 1. If one copy of chromosome 1 contains the RhD gene and one does not, the individual still expresses the Rh factor and is considered Rh positive.
42. Rh disease can cause haemolytic disease of the foetus, which in severe cases can result in stillbirth from anaemia. This issue typically arises in second or subsequent pregnancies when a Rh negative mother is carrying a Rh positive foetus. In other words, the child inherits from its mother a copy of chromosome 1 in which the RhD gene is deleted or mutated and a copy of chromosome 1 from the father in which the RhD gene is present. Since the child possesses one functioning copy of the RhD gene, the child produces Rh factor, and is thus referred to as Rh positive.
43. When a Rh negative mother carries a Rh positive foetus, the foetus expresses the Rh factor on its red blood cells. During pregnancy and birth the mother may be exposed to foetal red blood cells expressing Rh factor. The mother mounts an immune response to Rh factor, which it identifies as foreign, and thus her immune system becomes sensitised to Rh factor. A Rh negative mother sensitised to Rh factor may mount an immune response destroying the red blood cells of a Rh positive foetus in subsequent pregnancies.
44. Rh disease can be prevented by using a non-invasive test to determine the Rh status of the foetus, and treating all Rh negative mothers carrying Rh positive foetuses during pregnancy and/or immediately after childbirth with anti-Rh factor antibodies (so called prophylactic anti-D), ensuring that any Rh positive foetal red blood cells are masked before an immune response can be raised against them by the mother’s immune system, hence preventing issues with subsequent Rh positive pregnancies.
Cytogenetic analysis of foetal cells
45. Cytogenetic techniques analyse the number and structure of chromosomes. It includes techniques such as karyotyping and fluorescent in situ hybridisation (FISH) which allow for chromosomal abnormalities such as trisomies to be identified by the visualisation of the chromosomes in foetal cells. The karyotype of an individual is the number and appearance of the chromosomes in the nucleus of its cells.
46. Karyotyping involves the staining of chromosomes with a dye to allow them to be seen under a light microscope. Individual chromosomes may be identified by their length, the position of the centromere (the part of the chromosome at which the two arms are joined), and the banding pattern on the chromosome arms. Karyotyping therefore allows chromosomal abnormalities, such as autosomal trisomies, to be identified due to the presence of additional chromosomes. Karyotyping can also be used to identify disorders arising from loss or translocation of large sections of chromosomes (usually greater than 2 Mb).

47. FISH uses fluorescently-labelled single-stranded DNA probes designed to complement and bind to the portion of the gene of interest. The probe binds to complementary sequences on specific chromosomes, thereby allowing these complementary sequences of interest to be visualised by fluorescence microscopy. The presence of trisomy 21 may therefore be detected by use of a chromosome 21-specific probe. The presence of three fluorescent spots in the foetal cell, instead of the two which would be present in a normal (diploid) cell, indicates trisomy. This procedure is normally carried out on a number of cells from a sample to ensure a repeatable result.

Chorionic Villus Sampling
48. Chorionic Villus Sampling (CVS) is a method for the collection of placental cells that are likely to have the same karyotype as the foetus. The sample is collected from the chorionic villus of the placenta, either: (i) using a catheter which is inserted through the vagina and cervix to reach the placenta (transcervical CVS); or (ii) using a needle which is inserted through the abdomen of the mother into the placenta under the guidance of ultrasound (transabdominal CVS). After collection, checks are carried out in order to ensure that the cells are of chorionic villi and not of maternal tissue, and then the cells are cultured and subjected to cytogenetic analysis (see section L, above).

49. CVS is typically carried out during the first trimester of pregnancy. The risk of pregnancy loss arising from CVS is often quoted to patients at the upper limit of the range, approximately 1%.
Amniocentesis
50. Amniocentesis involves the collection of amniotic fluid, which contains foetal cells, using a needle which is inserted through the abdomen and uterus into the amniotic sac under the guidance of ultrasound. After collection, the foetal cells are cultured and subjected to cytogenetic analysis.
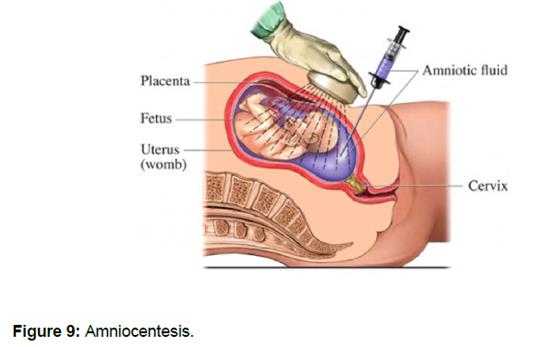
51. Amniocentesis is typically carried out during the second trimester of pregnancy. The risk of pregnancy loss arising from amniocentesis is often quoted to patients at the upper limit of the range, approximately 1%.
Foetal blood and tissue sampling
52. Foetal blood and tissue sampling involves the collection of foetal blood or tissue samples. Foetal blood samples may be collected using a needle which is inserted either into the umbilical cord or into the intrahepatic vein of the foetus under guidance of ultrasound. Foetal tissue samples, including skin, lung, liver and kidney samples, may be obtained using foetal biopsy techniques under either foetoscopic or ultrasonic guidance.
53. Foetal blood and tissue sampling can be carried out during the second trimester of pregnancy, but is typically avoided in practice due to the high risk of pregnancy loss, and because diagnoses can generally be made using amniotic fluid or CVS material.
Foetal cell-free DNA
54. In 1997 Professor Dennis Lo and colleagues discovered that the blood plasma and serum of pregnant women contains both maternal and foetal cell-free DNA (Lo et al , “Presence of fetal DNA in maternal plasma and serum”, Lancet , 350, 485-87 (1997). The following year Prof Lo’s group showed that foetal cell-free DNA was present in much higher quantities in maternal plasma and serum than were foetal cells in maternal blood and that foetal cell-free DNA could be detected as early as the seventh week of gestation and increased in concentration thereafter (Lo et al , “Quantitative Analysis of Fetal DNA in Maternal Plasma and Serum: Implications for Noninvasive Prenatal Diagnosis”, Am J Hum Genet , 62, 768-775 (1998), “Lo 1998”). These findings opened up the possibility of using foetal cell-free DNA for non-invasive prenatal diagnosis of foetal genetic traits.
55. In October 2003 clinical applications based on analysing foetal cell-free DNA focused on the qualitative detection of paternally-inherited foetal DNA not present in the maternal genome. Such applications included gender testing in pregnancies at risk of X-chromosome-linked disorders by detection of Y chromosome sequences, identification of pregnancies at risk of RhD haemolytic disease by detection of the RhD gene in the plasma or serum of RhD-negative mothers, diagnosis of human leukocyte antigen (HLA)-linked diseases, such as congenital adrenal hyperplasia, by detection of foetal HLA genes and detection of diseases resulting from paternally-inherited mutations, such as β-thalassaemia.
Polymerase chain reaction
56. The polymerase chain reaction (PCR) is a technique by which a DNA sequence can be “amplified” (i.e. multiple copies of that sequence can be generated) from a template DNA sequence. PCR uses a heat-stable DNA polymerase (which is an enzyme that produces new DNA strands) and short single-stranded DNA primers which are required for the initiation of DNA synthesis. In order to amplify the target sequence, the reaction takes place in the presence of an excess of free dNTPs (deoxyadenosine-triphosphate, deoxycytidine-triphosphate, deoxyguanosine-triphosphate, and deoxythymidine-triphosphate) in the reaction mixture. Deoxynucleotides are incorporated by the DNA polymerase in the generation of the new DNA strands, with the liberation of a pyrophosphate molecule.
57. PCR applications use thermal cycling, whereby the following steps constitute one cycle of PCR and are repeatedly carried out:
i) denaturation step: the reaction sample is heated to cause DNA melting, whereby the DNA strands separate to produce single-stranded template DNA molecules;
ii) annealing step: the reaction sample is cooled to allow annealing/binding of the DNA primers to regions of the single-stranded template DNA which are complementary in sequence to the primers; and
iii) elongation step: the temperature of the reaction sample is raised to a temperature at which the DNA polymerase is active and the primers are extended by incorporation of one nucleotide after another in a template dependent manner resulting in DNA synthesis of a new DNA strand complementary to the DNA template strand.
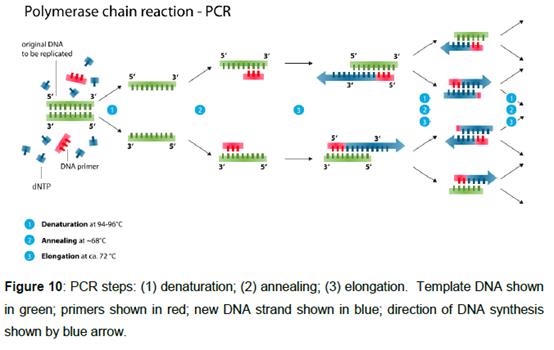
58. The amplification of new DNA strands is theoretically exponential (doubles with each cycle), as each new strand forms a template for the next round of synthesis. However, in reality, this will not be the case because non-optimal reaction conditions will mean that the efficiency of the PCR reaction is typically less than 100%. The efficiency of the PCR reaction will vary depending on many factors, including the annealing and extension temperatures, the polymerase and buffer conditions (ionic concentrations).
59. DNA primers must be designed to bind to opposite DNA strands and flank the target of interest and initiate synthesis of a new DNA strand complementary to the target sequence of each template strand. These primer pairs are commonly referred to as the forward and reverse primers.
Quantitative PCR
60. Quantitative PCR (qPCR) is a term used to describe the use of PCR to quantify nucleic acids. It can be accomplished using various procedures, one of which is real-time quantitative PCR. Real-time quantitative PCR involves a PCR during which accumulation of the amplified PCR product is monitored by measuring a signal created by either fluorescent dyes (such as SYBR Green) or fluorescent probes (such as TaqMan probes) in the reaction sample to generate an amplification curve. I shall describe qPCR in more detail below.
Gel electrophoresis
61. Gel electrophoresis is a technique for the analysis of a mixture of molecules (such as proteins or DNA fragments), which involves the use of an electrical current to draw them through a solid gel matrix, typically composed of agarose or polyacrylamide. The rate at which the DNA fragments pass through the gel depends on various factors, including their length, with shorter DNA fragments passing through the gel more quickly than larger fragments. The gel may be stained with a dye such as ethidium bromide, which fluoresces under ultraviolet light when intercalated with DNA, thus allowing the DNA fragments in the gel to be visualised. The size of the DNA fragments in the sample may be determined by use of a reference ‘ladder’ containing a mixture of DNA fragments of known sizes (see Figure 11 below). Gel electrophoresis, followed by excision of one or more regions of the gel and extraction of the DNA contained within the excised region(s), may be used to isolate DNA fragments of particular sizes.

Centrifugation
62. Centrifugation is a technique which allows particles in a solution to be separated on the basis of their size, shape and density by subjecting the solution to centrifugal force through spinning in a centrifuge. The correlation between the size and density of a particle and the rate at which it will separate from the mixture under centrifugal force allows particles of different size and density to be separated by applying different degrees of centrifugal force by varying the speed of the centrifuge. The rate of centrifugation is typically specified either in terms of revolutions per minute (RPM) or in terms of the centrifugal force applied to the sample measured in gravitational units (g).
The Patent
63. The Patent is a refreshingly short document. The specification begins at [0001] by referring to the finding (by Prof Lo) that the blood of a pregnant woman contains extracellular DNA from the foetus which can be detected in maternal plasma or serum, and that this can be used to detect foetal genetic loci which are absent from the maternal genome. At [0002] the specification notes that the determination of more complex foetal genetic loci is more difficult because the major proportion (generally greater than 90%) of extracellular DNA in the maternal circulation derives from the mother. This “vast bulk” of maternal circulatory extracellular DNA makes it difficult, if not impossible, to detect certain foetal genetic alterations.
64. The specification then introduces the invention in the following manner:
“[0003] An examination of circulatory extracellular fetal DNA and circulatory extracellular maternal DNA in maternal plasma has now shown that, surprisingly, the majority of the circulatory extracellular fetal DNA has a relatively small size of approximately 500 base pairs or less, whereas the majority of circulatory extracellular maternal DNA in maternal plasma has a size greater than approximately 500 base pairs. Indeed, in certain instances the circulatory DNA material which is smaller than approximately 300 base pairs appears to be almost entirely fetal. Circulatory extracellular fetal DNA in the maternal circulation has thus been found to be smaller in size (approximately 500 base pairs or less) than circulatory extracellular maternal DNA (greater than approximately 500 base pairs).
[0004] This surprising finding forms the basis of the present invention according to which separation of circulatory extracellular DNA fragments which are smaller than approximately 500 base pairs or less provides a possibility to enrich for fetal DNA sequences from the vast bulk of circulatory extracellular maternal DNA.
[0005] This selective enrichment, which is based on size discrimination of circulatory DNA fragments of approximately 500 base pairs or less, leads to a fraction which is largely constituted by fetal extracellular DNA. This permits the analysis of fetal genetic traits including those involved in chromosomal aberrations (e.g. aneuploidies or chromosomal aberrations associated with Down’s syndrome) or hereditary Mendelian genetic disorders and, respectively, genetic markers associated therewith (e.g. single gene disorders such as cystic fibrosis or the hemoglobinopathies), the determination of which had, as mentioned above, so far proved difficult, if not impossible. Size separation of extracellular fetal DNA in the maternal circulation thus facilitates the non-invasive detection of fetal genetic traits, including paternally inherited polymorphisms which permit paternity testing.”
65. Having acknowledged two items of prior art, and set out a consistory paragraph, the specification continues:
“[0008] The sample-fraction thus obtained not only permits the subsequent determination of fetal genetic traits which had already been easily detectable in a conventional manner such as the fetal RhD gene in pregnancies at risk for HDN (hemolytic disease of the fetus and the newborn), or fetal Y chromosome-specific sequences in pregnancies at risk for an X chromosome-linked disorder such as hemophilia, fragile X syndrome or the like, but also the determination of other, more complex fetal genetic loci, including but not limited to
- chromosomal aberrations (e.g aneuploidies or Down’s syndrome) or hereditary Mendelian genetic disorders and, respectively, genetic markers associated therewith (e.g. single gene disorders such as cystic fibrosis or the hemoglobinopathies);
and
- fetal genetic traits which may be decisive when paternity is to be determined.
[0009] Such determination of fetal genetic traits can be effected by methods such as, for example, PCR (polymerase chain reaction) technology, ligase chain reaction, probe hybridisation techniques, nucleic acid arrays (so-called ‘DNA chips’) and the like.”
66. There are two examples. Example 1 is described at [0011]-[0021]. In summary, this reports the following study. Seven women with third trimester pregnancies with a male foetus were recruited. Blood samples were collected and double-centrifuged (first at 1600 g for 10 minutes and then the supernatant was removed and spun at 16,000 g for 10 minutes). DNA was extracted from the plasma sample and precipitated. This DNA was then separated using gel electrophoresis, following which the gel was cut into pieces according to size markers. The resulting pieces of gel contained fragments of lengths 90-300 bp (base pairs), 300-500 bp, 500-1000 bp, 1000-1500 bp, 1500 bp-23 kb and greater than 23 kb. DNA was purified from the gel pieces. Finally, qPCR was used to quantify foetal DNA (using the SRY gene, amplicon size of 78 bp) and total DNA (using the GAPDH gene, amplicon size 97 bp) in each gel piece.
67. Results from five pregnancies are presented in Table 1 as follows:

68. The second column sets out the percentages of foetal DNA in each piece of the gel. The third column sets out the percentages of maternal DNA in each piece of the gel. The specification explains at [0021] that the figures in the second and third columns are the median values of the percentages and, in brackets, the ranges.
69. The specification comments on these results as follows:
“[0019] Table 1 shows that in the five pregnancies examined, DNA fragments originating from the fetus were almost completely of sizes smaller than 500 base pairs with around 70 % being of fetal origin for sizes smaller than 300 base pairs.
[0020] These results demonstrate that free DNA of fetal origin circulating in the maternal circulation can be specifically enriched by size separation of the total free DNA in the maternal blood. Depending on the downstream application the DNA size chosen for the enrichment of fetal DNA will be smaller than 300 or smaller than 500 bases.”
70. The specification does not show what the foetal fraction was before the size separation. Assuming that it was within the range published in Lo 1998 for third trimester pregnancies of 2% to 11%, the skilled reader would understand that the foetal fraction had been enriched in both the <300 bp and 300-500 bp fractions (and hence in the ≤500 bp fractions taken together).
71. The data do not show the absolute levels of DNA in each gel slice. Accordingly, it is common ground that the skilled reader would appreciate that the statement that “ DNA fragments originating from the fetus were almost completely of sizes smaller than 500 base pairs” is not actually established by the data presented.
72. Example 2 is an example of performing detection of genetic markers on plasma DNA fractionated on a gel as in Example 1. Table 2 reports results from PCR of a microsatellite (short tandem repeat) marker on chromosome 21. This is a locus where a sequence of four nucleotides is repeated over and over again, but with the number of repeats differing (in a heritable way) between different copies of a chromosome. In this instance, the mother’s two copies of chromosome 21 have lengths of 232 bp and 234 bp, and the foetus has inherited the 232 bp allele from the mother and a 228 bp allele from the father.
73. The results in Table 2 show that the foetal alleles could not be detected in the plasma DNA before size-separation, but the uniquely foetal allele of 228 bp could be detected in the size-separated fractions from gel slices containing DNA of either <300 bp or 300-500 bp.
The claims
74. The Claimants have applied unconditionally to amend the Patent to delete from claims 8 and 17 uses and processes for detecting chromosomal aberrations, and delete entirely claims 10, 11, 19 and 20, where the chromosomal aberration is an aneuploidy, and specifically one associated with Down’s syndrome. The reason for this is that the Claimants accept that those claims and parts of claims are invalid on the ground of insufficiency.
75. The remaining issues can all be determined by reference to claim 1, which is in the following terms:
“A fraction of a sample of the blood plasma or serum of a pregnant woman in which, as the result of said sample having been submitted to a DNA extraction, followed by a size separation, of the extracellular DNA, the extracellular DNA present therein substantially consists of DNA consisting of 500 base pairs or less.”
The skilled team or person
76. The Claimants contend that the Patent is addressed to a skilled team consisting of (i) a clinician qualified in obstetrics and gynaecology specialising in prenatal diagnosis and treatment and (ii) a human molecular geneticist with experience in the use of a range of standard techniques such as qPCR. The Defendants contend that the Patent is addressed to a skilled person with experience of working in a laboratory to detect genetic sequences in maternal plasma or serum with a view to providing clinical diagnoses and devising tests for this purpose. It is common ground that the differences between these formulations are of little significance and that, either way, the skilled team or person would have experience in carrying out genetic testing on maternal plasma or serum with view to clinical diagnosis
77. For what it is worth, Prof Hogge’s evidence was that all the leading groups in the field included a clinician whose involvement would have been in the clinical applications of the testing, and in my view this evidence fits with the specification of the Patent. Accordingly, I agree with the Claimants that the Patent is addressed to a team which includes a clinician. Nevertheless, since the remaining issues in the case lie mainly within the realm of the geneticist’s expertise, I shall for convenience refer to the skilled person.
Common general knowledge
78. There is little dispute as to the common general knowledge of the skilled person. In addition to the matters set out in the technical background section of this judgment, I find that it included the following.
Cell-free DNA
79. The cellular origin of cell-free DNA was poorly understood in October 2003, and the cellular processes by which maternal and foetal DNA were released into the maternal circulation were unknown. The release of foetal cell-free DNA was thought most likely to be from dying cells, with apoptosis thought likely to be the mechanism accounting for the majority of cell-free DNA (partly because it was known that a substantial degree of apoptosis occurred at the placental interface between the foetus and the mother), with cell death from necrosis as a possible contributor as well.
80. If asked, the skilled person’s expectation would have been that maternal and foetal cell-free DNA had a common origin, but there was no way of knowing. Similarly, the assumption would have been that the maternal and foetal cell-free DNA was of the same length, but this was something that the skilled person would have simply assumed without turning their mind to it. From a molecular biologist’s perspective, there was no reason to think that maternal and foetal DNA would be degraded differentially: both necrotic and apoptotic DNA would be degraded by nucleases in a continuing process, with even apoptotic DNA starting at a large size of 50-200 kb.
Extraction of cell-free DNA
81. The buffy coat contains mainly maternal white blood cells (leukocytes and thrombocytes), so it was known to be important not to disturb the buffy coat when removing the plasma or serum layer. Plasma and serum samples that were used for non-invasive prenatal diagnosis were nevertheless known to contain cell-free maternal DNA that was not of circulatory origin, but to have been released by lysis of maternal cells during subsequent handling and treatment. A precaution which was thought to be desirable in order to remove residual maternal cells from the plasma or serum, since release of DNA from these maternal cells could otherwise reduce the foetal fraction, was to perform a double centrifugation of the plasma/serum (first at low speed and then at high speed).
82. It was known that serum (as opposed to plasma) samples inevitably contained DNA that was not cell-free when the blood was in circulation, no matter what the centrifugation procedure. This is because DNA was released from blood cells ex vivo during the clotting process.
Efforts to improve detection of foetal cell-free DNA
83. While tests based on the presence/absence of a sequence not present in the mother (e.g. a Y chromosome sequence or RhD) had advanced to a good accuracy of detection by October 2003, testing for more subtle genetic differences still suffered from the problem caused by the background of maternal DNA. It was well known that Lo 1998 had shown that the mean foetal DNA fraction (i.e. the proportion of cell-free DNA that was foetal) rose from about 3% in early pregnancy to about 6% in late pregnancy, albeit varying widely in individual cases.
84. The maternal background was known to be problematic because it could give rise to non-specific amplification products, reducing the overall specificity of the assay. This was due to “mis-priming”, i.e. primers annealing to the wrong sequence present in the maternal DNA, which was particularly likely to occur when the target allele was similar to DNA in the maternal background, but could cause problems more generally, especially where there was a high level of maternal background.
85. Various attempts had been made to overcome the problem of the maternal background and to improve the reliability of foetal cell-free DNA detection, but there had been little success. The avenues that had been, or were being, explored in October 2003 included:
i) adopting best practice in handling and processing samples, by centrifuging without too much delay, and by using a double centrifugation, in order to try to prepare as pure a plasma sample as possible;
ii) mass-spectrometry coupled with a sensitive PCR technique to detect single nucleotide differences, which did not involve enriching or altering the source material;
iii) the (now discredited) suggestion of adding formaldehyde after blood draw to prevent cell lysis, which was a way of trying to avoid maternal DNA increasing in the sample, as opposed to reducing what was already there;
iv) exploiting differential methylation to identify a foetal allele as distinct from a maternal allele even though their sequences were the same, which did not involve any enrichment of foetal DNA; and
v) trying to find ways of enriching the foetal material in the maternal plasma or serum sample compared to the maternal DNA.
86. Apart possibly from the formaldehyde work, these approaches came from well-known and high-powered teams, and there were a number of other teams operating in the field over the period from 1997 to October 2003. Everybody in the field was generally interested in improving the detectability of foetal DNA, and the skilled person would have been on the lookout for any method of enriching the foetal cell-free DNA in a plasma or serum sample . However, nobody had ever suggested the possibility that there could be physical differences between the DNA fragments in circulation that would enable the maternal and foetal fragments to be separated after blood draw and DNA extraction.
Quantitative PCR
87. General . Quantitative PCR measures the increase in fluorescence after each PCR cycle in order to generate an amplification curve, from which the concentration of DNA in the original sample can be estimated from the exponential phase of the reaction. This is achieved by reference to the number of PCR cycles required to achieve a specified threshold level of fluorescence.
88. The threshold fluorescence level is set to a point in the exponential phase of the reaction (above the level of background noise but below the amplification plateau) when amplification is being performed most efficiently. The PCR cycle number at which the fluorescence signal reaches the chosen threshold is referred to as the cycle threshold (C T ). When the quantity of DNA of interest in the sample is high, the threshold level of fluorescence will be reached quickly and the C T value will therefore be low. Conversely, when the quantity of DNA of interest in the sample is lower, the threshold level of fluorescence will be reached more slowly and the C T value will therefore be higher. This is illustrated in the following graph, in which the C T value for the red sample is 24 while the C T value for the green sample is 25.
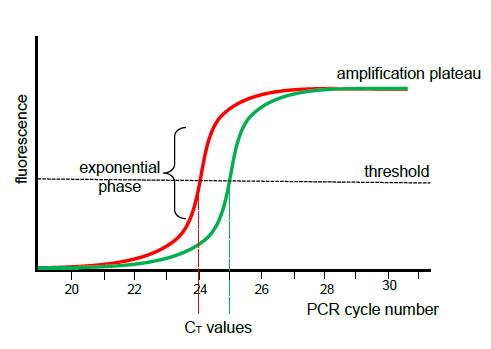
89. During the exponential phase of a qPCR reaction, there is a theoretical two-fold increase in the concentration of DNA every cycle and a corresponding doubling of the fluorescence signal .
90. Fluorescent reporters . As briefly noted in paragraph 60 above, there are various fluorescent reporters that can be used in qPCR to monitor the amount of product amplified as the reaction progresses. Some are non-sequence-specific dyes, such as SYBR Green. These fluoresce when bound to any double-stranded DNA (whether it be the intended amplification product or not), by intercalating between the base pairs. Such intercalating dyes will therefore fluoresce as a result of non-specific PCR products and primer dimers. The difficulties with SYBR Green could be avoided in a well-designed study, but it was not standard practice to do so in 2003.
91. Other qPCR reporters are sequence-specific, including probe hybridisation methods that can use two modified oligonucleotide probes which, when bound to DNA next to each other, result in fluorescence.
92. Amplification efficiency . One of the key parameters of qPCR reactions is the amplification efficiency, which may be estimated from a standard curve. To make a standard curve, qPCR is conducted on a series of known amounts of the DNA of interest, using the chosen primers and detection method (whether a dye or probe). This generates a series of amplification curves, each corresponding to known quantities of the sequence, which are then used to generate the standard curve for that sequence. The use of standard curves calibrates for potential differences in efficiency between different assays of interest, thus allowing copy numbers or concentrations for different samples to be determined and so compared.
93. Due to the exponential nature of qPCR, small differences in amplification efficiency between different targets or samples can result in large differences when amplified through multiple PCR cycles.
94. The efficiency of qPCR reactions is affected by a number of experimental parameters, including length of the amplicon, choice of polymerase, concentration of magnesium chloride (which acts as a cofactor to polymerase during the reaction), primer design and sample quality.
95. It was well known that, in general, the longer the length of the amplicon, the less efficient the qPCR reaction, and therefore PCR primers should be designed such that the amplicon length is as short as reasonably possible, typically in the range 100-150 bp.
96. Reference genes . In gene expression studies, it was standard to use a reference gene in qPCR. The same amplicon would be used across all of the samples being tested to normalise results, in order to control for factors that affected mRNA transcript level and to control for problems with reproducibility of PCR. A housekeeping gene was usually chosen as the reference gene for which expression levels were relatively constant.
97. Sensitivity and specificity . In a PCR reaction, sensitivity and specificity are intimately connected. If sensitivity can be increased (by increasing the amount of target sequence), it gives the freedom to improve the specificity by increasing the annealing temperature to reduce non-specific background amplification at the expense of some sensitivity.
98. Statistics . As with most analytical techniques, statistical information and analysis is essential in order to determine whether an apparent difference in qPCR results represents a real difference.
99. Relative vs absolute quantification . The concentrations of two target DNAs can be determined by qPCR in a relative or absolute manner:
i) relative quantification depends upon a comparison of the C T values (ΔC T ) to calculate the relative difference between them;
ii) absolute quantification depends upon quantifying the concentration of each target by use of a standard curve.
100. Figures derived from absolute quantification can be expressed as a ratio, but this does not mean that the resulting ratio is a relative quantification.
101. Relative quantification is inherently less accurate than absolute quantification. The accuracy of absolute quantification depends, inter alia , on the reliability of the standard curve being used.
102. Standard curves . Small differences in amplification efficiency can result in large differences in quantification, since small changes in the slope of a standard curve can result in large differences over the many cycles of exponential amplification used in PCR. This was illustrated by Prof Lovett in his second report, where he showed that a small change in the slope of the standard curve (resulting in a change in amplification efficiency from 1.73 to 1.67) would result in 2.4-fold difference in quantification over 25 PCR cycles:
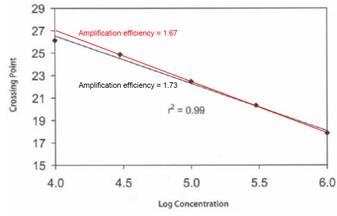
103. This effect can lead to a systematic error in absolute concentrations estimated using the standard curve, which cannot be rectified by making repeated measurements – all the concentrations could be out by, say, a factor of two.
104. Copy number . T he absolute copy number of a sample also affects repeatability (and reproducibility). High copy samples are more repeatable than low copy samples, with the coefficient of variation approximately doubling as the number of target molecules in the tube falls from 1000 to 100. Large differences in repeatability are also apparent between 10,000 copies and 1,000 copies.
105. It was known that foetal cell-free DNA was present in plasma at low copy number, with Lo 1998 using volumes of plasma that would contain fewer than 30 copies of SRY per reaction. Even if DNA from the entirety of the plasma from a late pregnancy blood draw of 10 ml, giving 5 ml plasma, was used, that would contain only five times the SRY copy number per ml figures given in Lo 1998, i.e. a median total of about 1,500 copies (range 384-3840).
106. Resolution. One area on which there was some disagreement between the experts concerns the resolution which could be achieved with qPCR in 2003. Although there was quite lot of evidence on this topic, in the end the differences between the experts were quite narrow. There was little disagreement that it was generally considered that the limit of detection was around a 1.5-fold difference, although Prof Lovett’s view was that this required both very careful experimental design using multiplex probes and appropriate statistical analysis. It was common ground between the experts that a 2-fold difference was regarded as reliably detectable, although Prof Lovett’s view was that this also required some care whereas Dr Daniels’ view was that it was routine.
107. Counsel for the Defendants submitted that the skilled person would not be surprised to see reports of observed 2-fold or 1.5-fold differences determined using qPCR, but they would know that that, without statistical analysis, the numbers should be taken as indicative rather than probative. I accept that subject to the rider that the skilled person would be more sceptical of a reported 1.5-fold difference than of a reported 2-fold difference.
Construction
108. There are three issues of interpretation of claim 1. It is common ground that the claim must be given a purposive interpretation. Claim 1 is a product-by-process claim, and it is also common ground that the process features are limitations on the scope of the claim for the purposes of infringement, obviousness and sufficiency (but not novelty): see Hospira UK Ltd v Genentech Inc [2014] EWHC 3857 (Pat) at [125]-[147] (Birss J).
Extracellular DNA
109. Claim 1 refers to “extracellular DNA”. There is no dispute that extracellular DNA means DNA that has been liberated from the cell. The Defendants contend that, in the context of claim 1, the skilled person would understand this refers to extracellular DNA of circulatory origin (i.e. DNA which was already extracellular when in circulation). The Claimants dispute this.
110. In my judgment the Claimants are correct on this point, for two reasons. Firstly, the specification of the Patent refers repeatedly to “circulatory extracellular DNA”, but the word “circulatory” does not appear in the claim. In my view the skilled person would conclude that this was a deliberate difference.
111. Secondly, it was common ground between Prof Hogge and Dr Daniels that the skilled person reading the Patent would be well aware that it was not possible to distinguish between extracellular DNA that had been extracellular in the circulation and any DNA that had become extracellular due to cell lysis after blood draw. The skilled reader would appreciate that all the extracellular DNA present in the sample would be subject to the separation process regardless of its origin. The skilled reader would also understand that the invention would be useful if it size-separated extracellular DNA derived from cell lysis after blood draw as well as extracellular DNA of circulatory origin. Accordingly, it would be contrary to the skilled person’s understanding of the purpose of the claimed invention to limit the claim to circulatory extracellular DNA.
Two-fold enrichment
112. The Defendants contend that claim 1 requires the removal of at least 50% of the maternal (circulatory) extracellular DNA that has been extracted from the plasma sample i.e. an enrichment of the proportion of foetal DNA by at least 2.0-fold. The Claimants contend that there is no such requirement.
113. In my judgment the Claimants are correct on this point. There is no such requirement anywhere in the Patent, let alone in the claims. The Defendants rely upon the first sentence of [0003]; but, as the Claimants point out, this sentence covers the situation where (say) 50.1% of foetal DNA and 49.9% of maternal DNA is 500 bp or less and hence there is scope for the invention to include even a slight degree of enrichment. The Defendants also rely upon the results of Example 1, which are summarised in [0019] as showing that “DNA fragments originating from the fetus were almost completely of sizes smaller than 500 base pairs”; but as the skilled reader would appreciate this is a single set of results from just five pregnancies. The specification does not suggest that all foetal cell-free DNA is smaller than 500 base pairs, and the skilled reader would appreciate that this was unlikely to be the case. The fact that the Defendants are driven to argue that enrichment must be at least 2.0-fold, and not merely 2-fold, confirms the lack of basis for their construction in the specification.
Size separation
114. Claim 1 requires the fraction substantially to consist of extracellular DNA of 500 bp or less following a size separation. In the alternative to their contention that the claim requires 2.0-fold enrichment discussed above, the Defendants contend that the claim covers a fraction derived from a sample of plasma or serum in which there was no extracellular DNA of greater than 500 bp originally. The Claimants’ primary case is that this is correct, because the claim is concerned with the end result of the process and not with whether the size separation step made any difference. In the alternative, the Claimants contend that the claims should be construed as being limited to fractions in respect of which the size separation step had some effect, and therefore as not covering fractions derived from samples in which there was no extracellular DNA of greater than 500 bp.
115. In my judgment the Claimants’ alternative construction is the correct one. In my view there can only be a size separation if there is something to separate. If there is no extracellular DNA of greater than 500 bp present in the sample originally, there is nothing to separate from the DNA of 500 bp or less.
Ikeda
116. Ikeda is a conference abstract which was published, in Japanese, prior to the 55 th Annual Congress of the Japanese Society of Obstetrics and Gynaecology, which took place in Fukuoka, Japan on 12-15 April 2003. Subject to one point which is discussed below, it is agreed that it is to be translated in English as follows:
“Frequency at which foetal DNA is present in maternal plasma: Difference by fragment length
Objective: It has been reported that foetus-derived free DNA is present in maternal plasma, and this has been confirmed to be unmistakably true in our study as well. In this study, we hypothesised that there would be more short-length DNA fragments, because foetal DNA is degraded by DNAse in maternal plasma.
Method: Using plasma samples from 9 women pregnant with boys, at 30 to 39 weeks' gestation, DNA was extracted with QIAamp DNA Blood Kit manufactured by QIAGEN. Quantitative PCR was performed using Roche's LightCycler. Primers targeting beta-globin (amplicon sizes of 110 bp and 196 bp: SYBR Green method) were prepared to estimate the amount of maternal DNA. In order to estimate the amount of foetus-derived DNA, we created our own primers and probes targeting SRY (amplicon sizes of 114 bp and 186 bp: Probe hybridization method), which exists only as a single copy on the Y chromosome. By studying these 4 regions, we examined DNA copy number and the percentage of the foetus-derived DNA at around 110 bp and around 190 bp.
Results: The frequency of SRY relative to beta-globin present in the maternal plasma was 19.6% for 114 bp and 9.8% for 186 bp. For overall beta-globin, 110 bp was detected 1.5 times more than 196 bp.
Conclusion: This study suggests the possibility that short-length DNA fragments are more prevalent. In the future, prenatal genetic diagnosis is likely to be carried out using foetus-derived DNA in maternal plasma, but it is possible that it will be more advantageous in terms of sensitivity and specificity to prepare PCR primers with the amplicon size as short as possible.”
The translation issue
117. The original Japanese contains a word, また or mata , at the beginning of the second sentence in the Results section which has not been translated in the English translation set out above. The Claimants contend that mata should be translated as “Furthermore” or “Moreover”. The Defendants contend that the translation is correct as it stands, and that no English word is required to translate mata . As counsel for the Defendants pointed out, the resolution of this issue is largely, if not entirely, irrelevant to the issue of how the skilled person would interpret Ikeda (although I think there is some force in the riposte of counsel for the Claimants that this is at least partly due to a shift in the Defendants’ case on the latter question). I shall nevertheless deal with it in some detail, because the points raised by it are of wider relevance.
118. It was common ground between counsel that what mattered was how the original Japanese would be understood. I am doubtful that this is correct, since the skilled person is located in the United Kingdom: see Generics (UK) Ltd v Warner-Lambert Co LLC [2015] EWHC 2548 (Pat), [2016] RPC 3 at [124]. It seems to me that it follows that the skilled person is deemed to read Ikeda in English translation. This point probably does not matter, however, since, even if it is the meaning of the Japanese that is determinative, an English court must rely upon a translation in order to appreciate that meaning. Either way, it is important that the translation should be as accurate as possible.
119. The next point to note is that translation is a form of expert evidence: see Sobrinho v Impresa Publishing SA [2015] EWHC 3542 (QB) at [3] and [23]-[24] and Umeyor v Ibe [2016] EWHC 862 (QB) at [38].
120. As Warby J pointed out in the first of these cases, it follows that the court’s permission is required to adduce such evidence under CPR Part 35. If a translation of a document is agreed, it is common for it to be relied upon without any formal order of the court giving permission, although in such a case the position could readily be formalised by an order giving permission for a single joint expert to give written evidence consisting of the agreed translation. (A similar approach could be applied to interpreters, while in the case of translations of affidavits and witness statements, it is arguable that the requisite permission is supplied by Practice Direction 32 paragraphs 10.2 and 23.2.) In the event of a dispute as to translation, however, permission must be sought and obtained to adduce expert evidence from translators. This was duly done in the present case.
121. As Warby J pointed out in both of the decisions cited above, it also follows that, in order for translation evidence to be admissible, the translator(s) must be appropriately qualified. I would add that, in the event of dispute, the qualifications of the rival translators will go to weight in the same way as the qualifications of any other expert do.
122. It is common practice for translations to be “certified”, that is to say, for the accuracy of the translation to be vouched for by the translator in a brief certificate. In my view it follows from the points discussed above that the certificate should be in the name of, and signed by, the translator who made the translation. In effect, it is a form of expert report. (In other words, a certificate signed only by a manager of a translation agency which employed the translator is not acceptable.) If it is anticipated that the translation will be agreed, then no doubt the full rigour of an expert’s report the form and content of which comply with Part 35, Practice Direction 35 – Experts and Assessors and the Guidance for the Instruction of Experts to Give Evidence in Civil Claims (including in particular details of the translator’s qualifications) may be dispensed with. In the event of dispute, however, reports which comply with these requirements will be needed. The reports in the present case did so.
123. Turning to the dispute in the present case, it is common ground that mata can be used as a noun, an adverb or a conjunction, that its meaning depends on how it is used and that in Ikeda it is used as a conjunction at the start of a sentence.
124. Prof Kornicki’s evidence was that, when mata is used as a conjunction at the start of the second of two sentences, its function is to signify a positive connection between the first and second sentences as opposed to a contrast. His opinion was that it was not correct to leave mata untranslated, since to do so would not convey the full meaning of the original Japanese. He explained that, when used as a conjunction, mata was used to indicate items that were either additional (“furthermore” or “moreover”) or parallel (“likewise” or “similarly”). I think he accepted that, in the context of Ikeda, the former meaning was more likely than the latter.
125. Prof Itoh’s evidence was that, even when used as a conjunction, mata is ambiguous and its meaning is highly dependent on context. He accepted that it could be used to imply a positive connection between two sentences, but said that it was often used as a filler or linking word which did not imply any positive connection between the two sentences, but simply an additional fact. His opinion was that, in the context of Ikeda, it was better not to translate the word. In the alternative, he considered that “also” or “in addition” would be an accurate translation. Although Prof Itoh considered that mata could be used as a contrastive conjunction, he did not suggest that it had that sense in Ikeda.
126. Although reference was made by the witnesses to dictionaries, these do not seem to me to assist, because the question is contextual. Similarly, although examples were produced of abstracts and other documents in which mata had been translated in various ways and also examples in which it had not been translated at all, I found the examples unhelpful since they merely generated disputes to the competence of the translators involved and as to the accuracy of those translations, and in any event the question is contextual.
127. In considering this issue, it seems to me that it is important to bear in mind that Ikeda is an abstract. It is common ground that it is probable that the authors were subject to a character limit (equivalent to a word limit in English). That being so, I consider it unlikely that they would have wished to waste two characters by including a word which was redundant. In those circumstances, it seems to me that it is not appropriate to leave mata untranslated. Furthermore, while I accept Prof Itoh’s evidence that it is not always strictly necessary to translate mata even in an abstract, I also accept Prof Kornicki’s evidence that fidelity to the original generally requires it to be translated.
128. That being so, it seems to me that there is little to choose between Prof Kornicki’s suggestion of “furthermore” or “moreover” and Prof Itoh’s suggestion of “also” or “in addition”. Considering the evidence as whole, however, I conclude that the most accurate rendition is “in addition”. Thus the second section of the results section should be translated as follows:
“In addition, for overall beta-globin, 110 bp was detected 1.5 times more than 196 bp.”
129. Turning to the impact of this on the skilled person’s understanding of Ikeda, Prof Lovett expressed the view that Prof Kornicki’s translation was consistent with the way he had interpreted Ikeda anyway, while Dr Daniels expressed the view that it made no difference which way Ikeda was translated. Neither expert was challenged on this in cross-examination.
What does Ikeda disclose?
130. There is a substantial dispute between the parties as to what, read through the eyes of the skilled person in 2003, Ikeda discloses. In brief summary, the Defendants contend that Ikeda discloses that foetal DNA fragments in maternal plasma are shorter on average than maternal DNA fragments, whereas the Claimants dispute this. For reasons that will appear, this issue is largely determinative of the issue of obviousness over Ikeda.
131. Prof Lovett supported the Claimants’ interpretation, while Dr Daniels supported the Defendants’ interpretation. As is common ground, it is for the Court to determine how the skilled person would interpret Ikeda, albeit guided by the expert evidence.
132. Ikeda must be interpreted as a whole. As is often the case with such disputes, however, it is convenient to analyse it in stages before reaching an overall conclusion.
The skilled person’s approach to Ikeda
133. Being a conference abstract, Ikeda is a very short document which is lacking in detail and in certain respects is unclear. The skilled person would also be aware that it had not been peer-reviewed. For these reasons, the skilled person would approach it with a degree of caution.
134. It is common ground that the skilled person is deemed to read a prior art document with care, and in that sense with interest, but not to assume that the document has any relevance to the problem the skilled person is addressing. This is an important point in the context of the present case. In these proceedings Ikeda has been subjected to sustained, detailed and high-powered forensic examination. That is not how it would have been read by the skilled person in 2003. I shall return to this point below.
Hindsight
135. It is also common ground that Ikeda must be interpreted without hindsight, that is to say, without knowledge of the invention disclosed and claimed in the Patent. The Claimants contend that this is an important point in the context of the present case, because the discovery which underpins the Patent that there is a size difference between foetal cell-free DNA and maternal cell-free DNA in maternal plasma and serum is one which is very difficult for someone who knows of it to put out of their mind. In those circumstances it is vital to read Ikeda without hindsight, yet hard to do so. I agree with this.
136. The Claimants further contend that Dr Daniels’ evidence was tainted by hindsight. As counsel for the Claimants pointed out, nowhere in his reports did Dr Daniels say that he was instructed, or attempted, to avoid hindsight when considering Ikeda despite the fact that he was very familiar with the Li 2004 and Chan 2004 papers discussed below. Moreover, in cross-examination, he accepted that he had had in mind his recollection that he personally had been aware at the filing date of the possibility of size separation (something that was plainly not common general knowledge). In re-examination he also indicated that he had taken into account his own work suggesting that foetal DNA fractions in maternal plasma were higher than had been reported in Lo 1998.
137. Counsel for the Defendants submitted that it had not been put to Dr Daniels that hindsight had affected the reasoning which underpinned his interpretation, nor explained how it could have. I do not accept this submission. In my view it was sufficiently put to Dr Daniels that his reading of Ikeda had been influenced by hindsight. Moreover, as I shall explain, I consider that hindsight is not only capable of affecting the way in which the document is understood, but did in fact colour Dr Daniels’ understanding of it.
The title
138. It is common ground that the skilled reader would start with the title. At one stage, the Defendants appeared to be contending that this suggests that the authors have found a difference in the fragment length of foetal DNA compared to maternal DNA. Rightly, this contention was not pursued in closing submissions. “Difference” after the colon must refer back to “Frequency” before the colon. Thus the title is indicating that the authors have found that foetal DNA present in maternal plasma differs by frequency of fragment length i.e. some fragment lengths are more frequent than others. There is nothing in the title to suggest a comparison between foetal DNA length and maternal DNA length. Indeed, there is no reference to maternal DNA at all.
The objective
139. It is common ground that the skilled reader would next read the objective. It is also common ground that the first sentence clearly refers to Prof Lo’s discovery. The second sentence is less clear: what is meant by the hypothesis that “there would be more short-length DNA fragments”? At one stage, the Defendants appeared to be contending the skilled reader would understand that this means more short-length fragments of foetal DNA than of maternal DNA. Rightly, this contention was not pursued in closing submissions. Again, there is no reference to maternal DNA fragments. Thus the skilled reader would understand that the authors hypothesised that there would be more short-length foetal fragments than long foetal fragments. This reading fits with the title.
140. Furthermore, the only mechanism mentioned in the objective is the degradation of foetal DNA by DNase in maternal plasma. As Dr Daniels accepted, there was no reason for the skilled person to think that DNase might act differentially on maternal, as opposed to foetal, DNA. Indeed, the mechanism of DNase degradation would be completely inadequate to explain the results, if the authors considered that their results indicated a physical distinction between foetal and maternal sequences in circulation.
141. Counsel for the Claimants put it to Dr Daniels in cross-examination that Ikeda’s reference to “short-length DNA fragments” would have been understood by the skilled person as referring to the lower end of a range that would have been considered to go up at least to kilobases and as covering the first few nucleosomal multiples of 160 bp, 320 bp and 480 bp. Dr Daniels accepted this without hesitation, and, although he retracted it in re-examination, that was in response to leading questions.
142. Counsel for the Defendants submitted that this was a new point which was not supported by the evidence of Prof Lovett, who had proceeded on the basis that “short-length” referred to fragments of between about 110 bp and about 190 bp. Counsel for the Claimants disputed this, but I consider that counsel for the Defendants is correct.
143. That is not the end of this point, however. The question is what the skilled reader would think that Ikeda was referring to at this stage of their reading of the document. Until the skilled reader gets to the method and results, the skilled reader has no basis for thinking that Ikeda is referring to fragments of between 110 and 190 bp. Ikeda does not define what is meant by “short-length DNA fragments” in the objective (or in the conclusion). In my judgment Dr Daniels’ evidence shows that, prior to reading the method and results, the skilled person would assume that Ikeda was talking about fragments of the order of 160 bp, 320 bp and 480 bp as opposed to longer fragments. But in any event, even if those particular values did not occur to the skilled reader, they would still think that Ikeda was drawing a contrast between short foetal DNA fragments and longer foetal DNA fragments. Moreover, for reasons that will appear, I do not consider that it matters what assumption the skilled person made before reading the method and results.
The conclusion
144. Dr Daniels agreed that the skilled person would be likely to look first at the title and objective and then the conclusion before delving into the technical detail of the method and results.
145. The conclusion tentatively suggests that “short-length DNA fragments are more prevalent”. I have discussed the meaning of “short-length” above. The conclusion then says that, in future, prenatal genetic testing is likely to be carried out using foetal DNA in maternal plasma. This would tend to confirm the skilled person’s assumption that the conclusion was one about foetal DNA. Finally, the conclusion suggests that it will be advantageous to use PCR primers with amplicons as short as possible. Both experts agreed that the suggestion of using short amplicons would be seen by the skilled person as sensible, but unremarkable since that was considered to be good practice anyway.
146. Two points follow from this. First, it is common ground that the authors do not state that they have discovered that foetal cell-free DNA is shorter than maternal cell-free DNA. As Prof Lovett put it, if the authors had thought that they had discovered this, they would have shouted it from the rooftops. Thus the Defendants are advancing an interpretation of Ikeda’s results which apparently did not occur to the authors themselves.
147. Secondly, as counsel for the Claimants submitted, in those circumstances, the skilled reader would not, when they turned to the method and results, have had their interest piqued or be expecting anything surprising. Moreover, they would assume that the method and results were consistent with the stated objective and conclusion. It follows that the skilled reader would not subject the method and results to sustained analysis looking for implications which the authors had not stated.
The method
148. The description of the method is brief and incomplete. Nevertheless, there is little dispute as to the meaning of most of the description, so far as it goes. The authors say that they carried out qPCR using primers targeting beta-globin with amplicon sizes of 110 bp and 196 bp to estimate the amount of maternal DNA and primers targeting SRY with amplicon sizes of 114 bp and 186 bp to estimate the amount of foetal DNA.
149. At this point it is necessary to explain the purpose and effect of using amplicons of different sizes. This was explained by Prof Lovett in his first report by reference to the following schematic diagram showing qPCR amplification of the beta-globin amplicons:
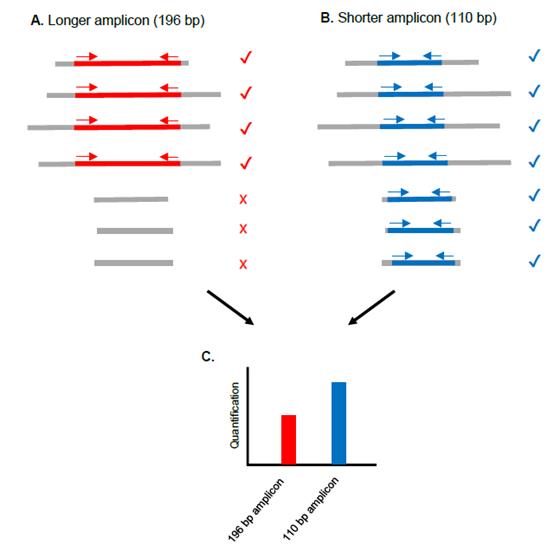
150. As shown in part A of the diagram, the PCR primers for the longer (196 bp) beta-globin amplicon (shown by the red arrows) will amplify DNA fragments (shown in grey) that contain the 196 bp target sequence (shown in red). As shown in part B, the PCR primers for the shorter (110 bp) amplicon (shown by the blue arrows) will amplify DNA fragments that contain the 110 bp target sequence (shown in blue). The ticks indicate where amplification of a fragment for a particular primer pair is possible; the crosses indicate where it is not. As shown in part C, since shorter-length beta-globin fragments (i.e. between 110 bp and 196 bp) are present, the level of quantification for the 110 bp amplicon (shown by the blue bar) is greater than the level of quantification for the 196 bp amplicon (shown by the red bar).
151. There is a dispute as to how the skilled reader would understand the last sentence of the method section, but it is convenient to consider this dispute together with the dispute as to the interpretation of the results. At first blush, however, the last sentence appears to be saying that the authors examined the percentage of foetal DNA at around 110 bp and around 190 bp. Thus, at least initially, this sentence would appear to be consistent with the title, objective and conclusion in focussing upon the relative proportions of different lengths of foetal DNA.
The results
152. The principal focus of the dispute concerns the results section, and in particular the first sentence. Although at one stage Dr Daniels drew a contrast between the first and second sentences, and indeed made a calculation on this basis, this point was not pursued (and any such reading would be somewhat undermined by my conclusion on the interpretation issue apart from anything else). Indeed, by the end of the trial, the Defendants were hardly relying upon the second sentence at all.
153. The crux of the dispute is this: what is the denominator in the two percentages reported in the first sentence? It is common ground that the text is not explicit in this respect, and that there are two possible interpretations. The Claimants contend that the skilled person would understand that the percentages were expressed by reference to the same denominator, namely the total amount of DNA of about 190 bp and above. The Defendants contend that the skilled person would understand that the percentages were expressed by reference to different denominators, namely the total amount of DNA of about 110 bp and above in the first case (19.6%) and the total amount of DNA of 190 bp and above in the second case (9.8%). The Defendants’ primary case is that, once the technical content of the document is fully understood, this is the only interpretation that makes sense of it. The Defendants’ secondary case is that the skilled person would at least realise that there were two possible interpretations of the document even if they were not sure which was correct.
154. The significance of the Defendants’ interpretation is that, on this reading, Ikeda is comparing the amount of foetal DNA of around 110 bp and above with the total amount of DNA of around 110 bp and above and comparing the amount of foetal DNA of around 190 bp and above with the total amount of DNA of around 190 bp and above, and finding that the first ratio (19.6%) is double the second ratio (9.8%). That would imply that there was more foetal DNA than maternal DNA present at short lengths (around 110 bp) compared to longer lengths (around 190 bp).
155. In considering this dispute, I begin with two points. The first is that there is nothing in the text of the first sentence to indicate to the reader that the two percentages are expressed by reference to different denominators. Absent any such indication, I consider that the skilled person would at least initially assume that they were expressed by reference to the same denominator. The second is that, given what is said in the title, objective and conclusion, I consider that what would catch the skilled reader’s eye would be the references to 114 bp and 186 bp, which the skilled reader would understand were the sizes of the amplicons created to estimate the amount of foetal DNA present. For these reasons, I consider that the skilled reader would at least initially assume that the first sentence was reporting a finding about the relative frequency of different lengths of foetal DNA.
156. In my judgment, this is where the question of hindsight becomes important. The Defendants’ primary case depends on the proposition that the skilled person would not simply make the assumptions that I have outlined, but would think more deeply about the technical sense of that reading of the document and, upon analysis, would conclude that it could not be correct. The Defendants’ secondary case depends on the proposition that the skilled person would at least recognise that it was possible that the percentages were based on different denominators even if they concluded that it was more likely that the percentages were based on the same denominator. I am not convinced, however, that, reading Ikeda without the benefit of hindsight, the skilled person would analyse the teaching of Ikeda in the manner postulated by the Defendants.
157. To my mind, it was a striking feature of Dr Daniels’ evidence that he said that the Claimants’ interpretation of Ikeda, that is to say, that the percentages were expressed by reference to the same denominator, had not even occurred to him until he read Prof Lovett’s first report. In my view this demonstrates that Dr Daniels’ reading of the document must have been affected by hindsight.
158. Turning to the evidence of Prof Lovett, he noted in his first report that it was possible that the percentages were being expressed by reference to different denominators, but expressed the opinion that it was most likely that they were being expressed by reference to the same denominator. In cross-examination Prof Lovett confirmed that he accepted that the skilled person would realise that it was possible that the percentages were being expressed by reference to different denominators, but maintained that they would conclude that it was most likely that the percentages were being expressed by reference to the same denominator. Counsel for the Defendants submitted that Prof Lovett’s key reason for expressing that opinion was coloured by Prof Lovett’s background in gene expression. I shall address that point below. Counsel for the Defendants nevertheless relied upon this evidence in support of the Defendants’ secondary case. Again, I shall return to that point below. At this stage, the point I wish to address is that Prof Lovett was also reading Ikeda with knowledge of the invention, although he recorded that he been instructed to try to avoid hindsight. Bearing that in mind, I am not persuaded that the skilled person reading Ikeda without hindsight would (as opposed to might) realise that the percentages could be referring to different denominators. Still less am I persuaded that the skilled person would engage in detailed analysis of Ikeda of the kind postulated by the Defendants.
159. I therefore conclude that the skilled person would simply take Ikeda at face value, would understand that it was all about the relative frequency of different lengths of foetal DNA and would assume that the percentages were expressed by reference to the same denominator even if it occurred to them that it was possible that they were expressed by reference to different denominators. On this reading, Ikeda would not suggest to the skilled person that there was a size difference between foetal DNA and maternal DNA.
160. In case I am wrong about that, however, I shall go on to consider what the skilled person would conclude if they analysed Ikeda more deeply. This requires consideration of a series of points.
161. Frequency of SRY relative to beta-globin . If the skilled person did analyse Ikeda more deeply, then the first question they would be likely to ask themselves was what Ikeda meant by the reference to “the frequency of SRY relative to beta-globin … for 114 bp and … for 186 bp [emphasis added]”.
162. As Dr Daniels accepted, the skilled person would start by assuming that the authors were presenting their results in a way which best met their objective. The skilled person would appreciate that the shorter SRY amplicon of 114 bp would catch foetal DNA fragments of that size upwards, whereas the longer SRY amplicon of 186 bp would catch foetal DNA fragments of that size upwards, but not smaller fragments. Thus, as noted above, the skilled person would think that the first sentence of the results was to do with a comparison between the relative frequency of foetal DNA fragments of 114 bp and upwards and 186 bp and upwards.
163. Dr Daniels agreed that, if the authors of Ikeda were trying to prove their hypothesis as set out in the objective, the logical thing to do would be to express the results for the two SRY amplicons relative to a common denominator. Ikeda says that the frequency of SRY is “relative to beta-globin”. As Prof Lovett noted, the reference to “beta-globin” could be a reference to either the shorter beta-globin amplicon (110 bp) or the longer beta-globin amplicon (196 bp), but his opinion was that it was more likely that the longer beta-globin amplicon was used as the common denominator. (This would be consistent with the use of the longer beta-globin amplicon as the denominator in the second sentence of the results.) Dr Daniels also agreed that, if the authors of Ikeda were trying to prove their hypothesis, and if they did compare the percentage detection of SRY at 114 bp and at 186 bp against the amount of beta-globin at 196 bp, then the authors’ conclusion that short-length foetal DNA fragments were more prevalent than longer length ones would have been a perfectly reasonable conclusion to reach. In other words, consideration of the reason for the comparison with beta-globin would reinforce the skilled person’s initial impression of the meaning of the first sentence of the results.
164. Single reference gene . The main reason given by Prof Lovett in his first report for concluding that the comparison was with the longer beta-globin amplicon was that it was normal to use a single reference gene in order that changes in the level of quantification could be directly compared between different genes. The Defendants advance a number of answers to this point.
165. First, they contend that, even if the skilled person expected a single reference gene to have been used, the design of the experiment shows that this is not what the authors were doing. This depends upon the Defendants’ interpretation of the disputed sentence in the method section, a point I shall consider below.
166. Secondly, the Defendants contend that a reference gene was understood to be an endogenous control with known characteristics, not a gene that the experimenter was studying. In this connection, as noted above, counsel for the Defendants submitted that it was significant that Prof Lovett’s background was in gene expression studies and he was used to seeing reference genes used in this way. It was therefore suggested that this had coloured his view. Prof Lovett vigorously disputed this suggestion when it was put to him. I am not persuaded that Prof Lovett’s evidence was coloured by his previous experience. As he pointed out, in the context of Ikeda, all that is needed in order for the comparison discussed above to be made is a common control, not a gene with a known level of expression. Counsel for the Defendants also relied upon the fact that, although Prof Lovett’s understanding was that use of a reference gene was common practice in the cell-free DNA field, he had not identified any other examples of this. I am unimpressed by this point, because what matters is what the skilled person reading Ikeda in 2003 would think that the authors had done. In my view they would regard the use of a common control as making sense.
167. Confidence in the data . The next question which the skilled person would be likely to ask themselves is what the significance of the apparent two-fold difference reported in the first sentence of the results was. There was quite a lot of evidence and argument about this.
168. It is common ground Ikeda gives little detail about the experimental methods used. Furthermore, there is no statement that replicates were performed, no ranges or error bars or standard deviations and no statistical information or analysis. So far as one can tell, the results appear to be simple averages of single measurements on each of the nine plasma samples. Dr Daniels agreed that the apparent two-fold difference reported in the first sentence of the results (let alone the apparent 1.5-fold difference reported in the second sentence) was reaching the point where the skilled person would wonder whether that was a real result or not. The Defendants therefore accept that the results are indicative, rather than probative. The Claimants contend that the point goes further than that, however.
169. First, the skilled team would be aware of the large biological variation between foetal DNA fractions in different samples. The spread of data shown by Lo 1998 for late pregnancy was 10-fold for SRY levels and nearly 30-fold for beta-globin, and the skilled person would have no reason to think that the SRY and beta-globin levels would vary in the same way in one individual. The consequence is that the biological variability would give rise to huge variation in the data across Ikeda’s nine samples.
170. Secondly, although Ikeda does not say how much plasma was used in each qPCR reaction, as explained above, the skilled person would be familiar with the use of volumes of plasma containing only very low copy numbers of SRY, such that the repeatability of qPCR results was compromised. There is nothing in Ikeda to suggest that steps have been taken to avoid this problem.
171. Thirdly, for beta-globin, a non-target-specific dye was used (SYBR Green), while for SRY a target-specific probe was used. Prof Lovett’s view was that this difference of detection methods would strike the skilled reader as an unusual and undesirable thing to do, since it would have introduced a further degree of variation into the results. (I will consider the reason which Prof Lovett surmised for this approach having been adopted below.) The Defendants rely upon a Roche technical note stating that its housekeeping gene standards can be used with a mix of detection methods (SYBR green for the target, probe hybridisation for the housekeeping gene). As Prof Lovett explained, however, the fact that it can be done does not mean it should be done. He said that it was not common, and he described it as a “red flag”. As counsel for the Claimants pointed out, there is no example in evidence where it has in fact been done, let alone published in a peer-reviewed journal.
172. Fourthly, the skilled person would note that Ikeda does not state whether relative quantification or absolute quantification was used.
173. Prof Lovett opined in his third report that the skilled reader would have thought it likely that Ikeda had used a relative quantification protocol rather than an absolute quantification method for two reasons. He conceded the first reason in cross-examination, but maintained the second, which is that absolute quantification is far more labour intensive than relative quantification. In Prof Lovett’s view the explanation for Ikeda’s use of the SYBR Green method for beta-globin was that the authors were unwilling to spend the time and effort needed to generate hybridisation probes and primers for beta-globin, which in turn made it unlikely that they had employed absolute quantification.
174. In his second report Dr Daniels relied upon the use of the expression “copy number” in the last sentence of the method section as indicating that absolute quantification was used. As Prof Lovett pointed out, however, copy number can be measured in an absolute or relative manner. I understood Dr Daniels to accept this in cross-examination. Instead, he expressed the view that, unless it was assumed that absolute quantification was used, Ikeda would not even have been worth reading.
175. The conclusion I draw from this evidence is that the skilled person would consider it likely that Ikeda had used relative quantification, which would further reduce their confidence in the significance of the results. That in turn would mean that they would not devote any further effort to analysing Ikeda.
176. Even if the skilled person assumed that absolute quantification was used, as explained above, the skilled person would know that the amplification efficiency for a single amplicon could easily be out by enough to make its quantification wrong by a factor of two. Prof Lovett explained that this gives rise to another important advantage of using a common reference gene, in that it reduces the number of variables in the experiment. Given the real difficulties with accurately assessing the amplification efficiency for any given amplicon, it would make no sense to use different denominators. As Dr Daniels accepted, using a different denominator (i.e. a different beta-globin amplicon) for each of the two SRY amplicons would mean that the first sentence of Ikeda’s results section would depend on the results for four different amplicons and hence upon the combined errors for each of those four amplicons.
177. The conclusion I draw from this evidence is that, even if the skilled person considered that absolute quantification had been used, they would conclude that the results could only be meaningful if they were expressed by reference to the same denominator. This would tend to reinforce their assumption that that was how the results were expressed.
178. In conclusion, consideration of the significance of the apparent 2-fold difference reported in the first sentence of the results would either lead the skilled person to conclude that it was unreliable, in which case they would devote no further effort to analysing Ikeda, or would reinforce the skilled person’s assumption that the results were expressed by reference to the same denominator.
179. The design of the experiment and foetal fractions . In closing submissions counsel for the Defendants placed at the forefront of his argument the way in which the Defendants contend that the skilled person would understand the experiment to have been designed. Before considering the detail of this contention, I would make two preliminary observations. The first is that the contention depends on an analysis of the method section of Ikeda, but that is the part of Ikeda which the skilled person would pay least attention to in the absence of something elsewhere in the document to pique their interest or which called for explanation. The second is that, for reasons that will appear, the contention pre-supposes that, contrary to what is suggested by the title, objective and conclusion, the authors of Ikeda were in fact intending to measure the relative sizes of foetal and maternal DNA. That is inherently improbable.
180. The key to the argument is Ikeda’s use of four primers, two for maternal DNA and two for foetal DNA, and the explanation which Ikeda gives for the use of the primers in the last sentence of the method section, and in particular the reference to the percentage of foetal DNA “at around 110 bp and around 190 bp”. The Defendants point out that both pairs of primers were around 110 bp and around 190 bp: 110 bp and 196 bp in the case of beta-globin and 114 bp and 186 bp in the case of SRY . Accordingly, the Defendants contend, the skilled person would conclude that what Ikeda had done was to design the experiment to compare the quantities detected by 114 bp SRY with 110 bp beta-globin (short:short) and to compare the quantities detected by 186 bp SRY with 196 bp beta-globin (long:long).
181. In evaluating this argument, it is important to understand two points which it is common ground the skilled person would appreciate if they thought about it. The first is that, although Ikeda states that beta-globin was used to estimate the maternal DNA, in fact it would have been a measure of the total DNA (maternal and foetal) because the gene is present on chromosome 11. It follows that, on Dr Daniels’ interpretation of the way in which the experiment was designed, what Ikeda was measuring were the short and long foetal fractions of the total short and long DNA fragments. A difference between the two would, of course, indicate a difference in the amount of short foetal DNA compared to short maternal DNA.
182. Secondly, the SRY gene is present in a single copy (because it only occurs on the Y chromosome), whereas the beta-globin gene is present in two copies (one on each copy of chromosome 11). It follows that a ratio of SRY:beta-globin is not equal to the foetal fraction of the total DNA. As Dr Daniels accepted, it would be necessary to double the SRY:beta-globin figure in order to calculate the foetal fraction.
183. Dr Daniels opined in his second report that the percentages which Ikeda reported in the first sentence of the results section (19.6 and 9.8) were foetal fractions, implying that Ikeda had already doubled the SRY figures to reach the percentages reported. This is contrary to what Ikeda says, however, which is that it is reporting the frequency of SRY relative to beta-globin. Dr Daniels accepted that his interpretation of the document required the authors to have misreported their results in the results section. This is highly improbable.
184. Furthermore, if the reported percentages were foetal fractions, the figure of 19.6% would be very high compared to the normal range known from Lo 1998, which in late pregnancy was from 2.3% to 11.4% with a mean of 6.2%, using similar amplicon sizes to Ikeda’s 110 bp. When this point was put to Dr Daniels, he revealed that he had taken into account his post-filing date knowledge that foetal fractions were often higher than reported in Lo 1998. He accepted that the skilled person without that knowledge would have found a foetal fraction of 19.6% to be a remarkable result. This reinforces the conclusion that the skilled person would think that the percentages reported were SRY:beta-globin figures and not foetal fractions.
185. If, on the other hand, the percentages reported were SRY:beta-globin figures, then the Defendants’ interpretation implies that the foetal fractions detected were 39.2% and 19.8%, which would be even more remarkable. Thus the Defendants’ interpretation requires Ikeda not only to have omitted to report in the conclusion that the authors had found a size difference which (without mentioning it in the objective) their experiment had been designed to detect, but also that they had found the presence of a much higher foetal fraction than had previously been reported (which would in itself make prenatal testing easier). This is to pile improbability upon improbability.
186. Although the Defendants contend that the language of the last sentence of the methods section is only consistent with their interpretation, I disagree with this. The Claimants do not dispute that the references to “around 110 bp” and “around 190 bp” are shorthand which avoids spelling out all four amplicon sizes. But the language used does not compel the conclusion that the short SRY amplicon was compared to the short beta-globin amplicon and the long SRY amplicon was compared to the long beta-globin amplicon. On the Claimants’ interpretation, Ikeda did compare the percentages of short and long foetal DNA, but by reference to the long beta-globin comparator. (Ikeda also compared the amount of short and long total DNA, which is reported in the second sentence of the results, but the skilled person would not think that the apparent 1.5 fold difference detected was significantly different to the apparent 2-fold difference between short and long foetal DNA.)
187. The Defendants also contend that the Claimants’ interpretation is inconsistent with Ikeda’s conclusion. This contention is based upon an analysis put to Prof Lovett in cross-examination of what would happen in Ikeda in a perfect experiment. In theory, if there are equal numbers of short (114-186 bp) foetal DNA fragments and long (186 bp and above) foetal fragments, then the observed difference in quantification between them in a perfect experiment would be 2-fold. This is because, as illustrated by Prof Lovett’s schematic diagram, the short (114 bp) SRY amplicon will pick up the long fragments as well as the short fragments, while the long 186 bp amplicon will just pick up the long fragments. It would follow that, if the reported 2-fold difference (19.6% vs 9.8%) is understood to be a comparison between short and long foetal fragments as measured by SRY relative to a common control, then that would not support Ikeda’s conclusion that short foetal fragments were more prevalent than long ones. Accordingly, the Defendants contend, the skilled person would conclude that Ikeda could not be comparing short and long SRY relative to a common control, but must be comparing them relative to short and long beta-globin respectively.
188. Prof Lovett accepted that this analysis was correct as a matter of theory, but disputed that it would occur to the skilled person reading Ikeda. In my judgment this is not an analysis that would occur to the skilled reader without hindsight. It is not even an analysis that, so far as I can see, was advanced by Dr Daniels in any of his three reports. Furthermore, even if the point did occur to the skilled reader, the skilled reader would still be confronted by (among other things) the problem that the Defendants’ interpretation requires the skilled person to conclude that, without mentioning the fact in their title, objective or conclusion, the authors had designed their experiment to detect differences between the lengths of foetal and maternal DNA fragments and had succeeded in doing so. In my view the skilled person, if they thought about this point at all, would be more likely to conclude that this was a flaw in the experimental protocol which the authors had not spotted. That would be consistent with (i) the authors’ erroneous reference to beta-globin measuring maternal DNA when in fact it measured total DNA, (ii) the use of the two different detection methods and (iii) the absence of any reference to the SRY gene being present as one copy whereas the beta-globin gene was present as two copies.
189. Finally, it remains for me to deal with a point made by the Defendants about Ikeda’s conclusion which I have not so far dealt with. This is that Ikeda’s reference to it being advantageous in terms of both sensitivity and specificity to use short amplicons points to the experiment being designed in the manner suggested by the Defendants. Prof Lovett did not accept this and nor do I. It may be the case that this conclusion is not fully supported by the results on the Claimants’ interpretation, but if so the skilled person would not find that surprising. But the point does not answer all the problems with the Defendants’ interpretation discussed above.
190. Overall, therefore, I conclude, that, even if the skilled person analysed Ikeda in more depth than I consider that they would, they would reach the same conclusion, namely that what Ikeda discloses is that short foetal DNA fragments may be more prevalent in maternal plasma than longer foetal DNA fragments.
Obviousness over Ikeda
191. As noted above, the obviousness of claim 1 over Ikeda depends almost entirely upon how Ikeda is interpreted. I do not understand the Defendants to contend that claim 1 is obvious if Ikeda is interpreted as the Claimants contend, as I have concluded it would be. The Claimants do not concede that claim 1 is obvious if Ikeda is interpreted as the Defendants contend on their primary case, but do not advance any serious case to the contrary. There is no dispute that the skilled person would have had a motive to find a method of enriching the amount of foetal cell-free DNA in a plasma or serum sample compared to the maternal background, or that carrying out a size separation test would only require routine techniques which would have been quick and easy to perform. Although, even on the Defendants’ case, the data in Ikeda are only indicative, rather than probative, of a size difference, that would give the skilled person sufficient expectation of success to warrant carrying out the experiment. Accordingly, I conclude that, on that hypothesis, claim 1 would be obvious. I do not understand the Claimants to dispute that, if claim 1 is obvious over Ikeda, then so are the remaining claims.
192. It only remains for me to deal with the Defendants’ secondary case that the skilled person would realise that a possible interpretation of the results section of Ikeda was that it was expressed by reference to different denominators, and that is sufficient to render claim 1 obvious over Ikeda. I do not accept this. In the first place, for the reasons explained above, I am not convinced that the skilled person would (as opposed to might) realise this. Secondly, even if the skilled person did realise this, I consider that the skilled person would conclude that that was not the correct interpretation of Ikeda. Once the skilled person has concluded that the correct interpretation of Ikeda is that it is all about the size distribution of foetal DNA, there is no room for an obviousness case based on the alternative interpretation. The only course that would be obvious, as the Claimants’ witnesses accepted, would be to repeat Ikeda with more rigour to see if the same results were obtained with statistical significance.
Insufficiency
193. The Defendants contend that the claims are invalid on the ground of insufficiency.
The law
194. The relevant law was recently reviewed by the Court of Appeal in Regeneron Pharmaceuticals Inc v Kymab Ltd [2018] EWCA Civ 671, [2018] RPC 14 at [208]-[249].
195. At [214]-[230] the Court reviewed the case law of the Boards of Appeal of the European Patent Office. Having cited with approval Jacob LJ’s summary i n Novartis AG v Johnson & Johnson Medical Ltd [2010] EWCA Civ 1039 of the heart of the test as being “Can the skilled person readily perform the invention over the whole area claimed without undue burden and without needing inventive skill?”, the Court added three points at [231]-[233]:
“231. First, it is not the law that a specification must necessarily enable the skilled person to make or perform all of the embodiments of a claimed invention. Were it otherwise, claims would be insufficient if they covered inventive improvements. But, as the decision in Polypeptide expression/Genentech I makes clear, in appropriate cases, a claim may embrace variants which may be provided or invented in the future and which achieve the same effect in a manner which could not have been envisaged without the invention.
232. Secondly, the assessment of insufficiency must be sensitive to the nature of the invention and the facts of the particular case. If the character of the invention is one of general methodology or is such that the invention is of general application then it may be permissible to claim it in general terms, even though the specification does not enable every way of arriving at its subject matter. Otherwise, as the Board explained in Modifying plant cells/MYCOGEN , no dominant patent could ever exist and each developer of a new method of arriving at that subject matter would be free of earlier patents. In many cases in the field of biotechnology, patent protection would then become illusory.
233. Thirdly, it is a general principle that the protection afforded by the claims must correspond to the technical contribution to the art made by the disclosure of the invention. The patentee is entitled to fair protection having regard to the nature and character of the invention he has described.”
196. The Court then turned to consider the English case law. At [238] the Court drew the following points from Biogen v Medeva [1997] RPC 1:
“i) The extent of the patent monopoly, as defined by the claims, must correspond to the technical contribution to the art its disclosure has made in order for it to be justified.
ii) The specification must enable the invention to be performed to the full extent of the monopoly claimed. But if the invention discloses a principle capable of general application, the claims may be in correspondingly general terms.
iii) If the patentee has found a new product which has a beneficial effect but cannot demonstrate there is a common principle by which that effect will be shared by other products of the same class, he will be entitled to a patent for that product but not for the class. But if he has disclosed a beneficial property which is common to the class, he will be entitled to a patent for all the products of that class even though he has not himself made more than one or two of them.
iv) There is more than one way in which the breadth of the claim may exceed the technical contribution to the art embodied in the invention. The patent may claim results which it does not enable, such as making a wide class of products when it enables only one of those products and discloses no principle which would enable others to be made. Or it may claim every way of achieving a result when it enables only one way and it is possible to envisage other ways of achieving that result which make no use of the invention.”
197. At [245] the Court drew the following points from the decision of the House of Lords in Kirin-Amgen Inc v Hoechst Marion Roussel [2004] UKHL 46, [2005] RPC 9 and the decisions of the Court of Appeal and the House of Lords in H Lundbeck A/S v Generics (UK) Ltd [2008] EWCA Civ 311, [2008] RPC 19 and [2009] UKHL 12, [2009] RPC 13:
“i) a principle of general application is simply an element of a claim which is stated in general terms;
ii) a claim containing such an element is sufficiently enabled if the skilled person can reasonably expect the invention to work with anything which falls within the general term; and
iii) a particular form of an element of a claim may improve the way the invention works and be inventive. However, the patent is not insufficient simply because the specification does not enable that improvement. It is still a way (albeit an improved way) of working the original invention.”
198. At [249] the Court expressed the view that its previous decision in Regeneron Pharmaceuticals Inc v Genentech Inc [2013] EWCA Civ 93, [2013] RPC 28 was consistent with the principles it had identified and re-iterated:
“A claim is not insufficient simply because it encompasses inventive improvements provided they embody the technical contribution the disclosure of the invention has made to the art.”
The Defendants’ case in outline
199. The Defendants contend that the claims made in the Patent are at odds with what it is now known about the size distribution of foetal cell-free DNA compared to that of maternal cell-free DNA. The Defendants say that the claim that the majority of circulatory extracellular maternal DNA is greater than 500 bp is simply wrong and that, although on average circulatory extracellular foetal DNA is somewhat shorter in length than circulatory extracellular maternal DNA, this difference is nowhere near as large as that reported in the Patent. In support of this contention, the Defendants rely upon the evidence of Prof Thierry. His opinion is that the data in the Patent were confounded by contamination from genomic DNA released by maternal cell lysis after blood draw due to poor sample handling.
200. The Defendants further contend that, because there is only a small size difference between foetal and maternal DNA, removal of circulatory extracellular DNA above 500 bp from a plasma or serum sample achieves very little, and certainly does not represent a generally applicable means of obtaining a technically useful class of product.
201. The Defendants argue this gives rise to a squeeze between construction (and hence infringement) and insufficiency: either the claims are construed as requiring a 2.0-fold enrichment of foetal DNA or else they are insufficient because their scope is broader than the technical contribution in fact made by the Patent. I have already dealt with the construction of the claims above. I must now address the factual basis for the Defendants’ insufficiency case before turning to the legal consequences of the facts. This requires consideration of three papers published by workers in the field since the filing date of the Patent.
Chan 2004
202. The best source of data on the size distribution of cell-free DNA fragments in evidence is a paper by Chan et al , “Size Distributions of Maternal and Fetal DNA in Maternal Plasma”, Clin Chem , 50(1), 88-92 (2004) (“Chan 2004”). In this paper Prof Lo’s group investigated the size distribution of plasma DNA in 31 pregnant women, 34 non-pregnant women and 16 men (as controls) using qPCR . They used six differently-sized amplicons targeting the SRY gene to estimate the foetal DNA and nine differently-sized amplicons targeting the leptin gene to estimate the maternal DNA. They concluded that foetal cell-free DNA is shorter than maternal cell-free DNA, and suggested (at page 92) that this different size distribution “may open up a possible way to enrich for fetal DNA by size fractionation of DNA extracted from the plasma of pregnant women”. On its face, therefore, Chan 2004 is broadly consistent with the Patent.
203. Prof Thierry did not criticise Chan 2004 in any of his reports, and instead put it forward as a reliable source of information. In his oral evidence, he described the research as “well done” and from a “great group”. He also said that Chan 2004 contained the best data using qPCR with regard to circulating DNA over 500 bp in pregnant women.
204. It is common ground that Chan 2004 does show that the majority of maternal cell-free DNA is greater than 500 bp in some cases, but that those cases are in the minority. Figure 1A compares the size distributions of DNA in pregnant (red) and non-pregnant (blue) women, while Figure 1B compares the size distributions of foetal (blue) and maternal (red) DNA:
205. Figure 1 shows that the median proportions of fragments amplified by (and hence at least as large as) primers for the 449 bp and 576 bp leptin amplicons in pregnant women are 30% and 21% respectively. Prof Thierry accepted an interpolated value of around 25% at 500 bp.
206. As is also common ground, however, the spread of data is large, with the 75 th percentiles being at around 55% and 43% for the 449 bp and 576 bp amplicons, respectively. The 75 th percentile at 500 bp would be somewhere between these two, in the region of 50%. At this size, Figure 1B shows that there is virtually no foetal cell-free DNA at all. Thus the majority of maternal cell-free DNA in pregnant women is above 500 bp in about 25% of cases, according to Chan 2004’s data.
207. Prof Lovett’s opinion was that Chan 2004’s Figure 1A shows that the existence of maternal cell-free DNA of greater than 500 bp in pregnant women is a real effect, not due to contamination. I accept this evidence, for the following reasons.
208. As noted above, Figure 1A shows the size distributions of plasma cell-free DNA from pregnant women (in red) and non-pregnant women (in blue). The text of the paper states that there was no statistically significant difference between the plasma DNA concentrations for the two groups. As Prof Thierry accepted, this suggests that the handling of the samples was carried out consistently between the two groups. Prof Thierry later suggested that the authors might have made a mistake so that there was in fact a difference between the handling of the two groups, but there appears to be no basis for this supposition.
209. As Prof Thierry also accepted, Figure 1A shows a statistically significant difference between the two groups for all amplicon sizes from 201 bp upwards, in each case with a p value of <0.001. The phenomenon of cell lysis after blood draw would apply equally to samples taken from both groups, however. Comparing the blue and red bars above 500 bp therefore shows that any effect of contamination by cell lysis in this data is marginal, even if all the DNA of these sizes in non-pregnant women represented contamination.
210. Taking the interpolated median figure of 25% for the proportion of maternal DNA of greater than 500 bp in pregnant women, and subtracting the median for non-pregnant women of 7%, leaves about 18% of the total maternal DNA that is greater than 500 bp and that cannot be due to contamination.
211. Prof Thierry’s only answer to this was to appeal to an unidentified source “in the literature” in which an unspecified experiment had showed a “mean, what you can find over 500 [bp], is 12%”. That figure, however, was for non-pregnant individuals. It is higher than that reported by Chan 2004 for the median of its corresponding cohort, which had a median of 7% of cell-free DNA larger than either 449 bp or 576 bp. In any event, Chan 2004 shows that the amount of cell-free DNA greater than 500 bp in non-pregnant women is significantly lower than for pregnant women. Prof Thierry has not done his own research on the effect of pregnancy on sequences of greater than 500 bp.
212. Based on the Chan 2004 data, Prof Thierry agreed that the proportion of total cell-free DNA over 500 bp in pregnant women would vary from close to zero to around 60-70% in any individual, while there would not be any significant quantity of foetal DNA in this size range.
213. The Claimants contend that Chan 2004 supports the invention of the Patent in that it shows that removing cell-free DNA of 500 bp from a plasma sample would be expected to increase the foetal fraction of cell-free DNA over and above the foetal fraction that existed in circulation. I accept this subject to the qualification that the data shows that, in some cases, this would have little effect.
Li 2004
214. Li et al , “Size separation of Circulatory DNA in Maternal Plasma Permits Ready Detection of Fetal DNA Polymorphisms”, Clin Chem , 50(6), 1002-1011 (2004) (“Li 2004”) is a paper by the inventors of the Patent (plus two others) and includes some of the same experimental results.
215. The authors credit Chan 2004 with discovering that foetal DNA fragments are generally smaller than maternal DNA fragments. The authors confirm this observation, focusing on the size distribution of larger maternal DNA. They do this using a combination of Southern blotting, agarose gel electrophoresis and qPCR. The results of the agarose gel electrophoresis and qPCR experiments are set out in Tables 1 and 2.
216. Li 2004 has been cited several hundred times since it was published as evidence of the difference in the size distribution between maternal and foetal extracellular DNA, and Prof Hogge was not aware of any publication that suggests that the results reported in Li 2004 are exaggerated or incorrect due to contamination of the samples.
217. Prof Thierry agreed that Li 2004 has been cited multiple times for the proposition that the lengths of fragments of maternal cell-free DNA in circulation are significantly higher than those of foetal cell-free DNA. It was nevertheless his opinion that the authors had not taken enough care about pre-analytical issues that can result in maternal contamination.
218. Table 1 of Li 2004 presents size data from six samples from third-trimester maternal plasma samples while Table 2 presents size data from eight samples from early pregnancy samples. Prof Lovett and Prof Thierry agreed that the samples in Table 1 appeared to be the same five samples as in Table 1 of the Patent with one extra sample:

219. The paper provides additional data on these samples that shows the distribution of the DNA across different gel slices (rather than providing only the distribution as between maternal and foetal DNA within each gel slice). It shows that, of foetal cell-free DNA, a median of about 70% is in the less than 300 bp gel slice while a median of about 24% is in the 300-500 bp gel slice.
220. For maternal cell-free DNA, Li 2004 shows that around half is likely to be in the 500 bp to 23 kb range: adding up the medians for the gel slices gives 51.6% of the total cell-free DNA above 500 bp for Table 1, and 45.3% for Table 2.
221. There was a dispute between the experts over the implications of the Southern blot in Figure 1 of Li 2004, which I reproduce below. This shows in lane 2 the presence of high molecular-weight DNA (around 23 kb) in the plasma of a pregnant woman at 13 weeks gestation, using a probe that binds to the Alu sequence that is present commonly throughout the genome. Lane 3 shows plasma from a non-pregnant woman.

222. The authors say (at page 1006):
“This examination also indicated that a substantial proportion of the circulatory DNA had a molecular size >10 or even >23 kb (Fig. 1). The presence of such high-molecular-weight DNA species cannot be attributed to the plasma sample being contaminated by maternal cells because we took extreme care to obtain cell-free plasma samples.”
223. The authors also say (at page 1008):
“With regard to the size distribution of total circulatory DNA, we determined that the pattern we had observed in pregnant women was very similar to that observed in samples taken from nonpregnant women as well as healthy male volunteers (Fig. 4). In none of these analyses were we able to detect large amounts of DNA with a molecular size greater than that indicated by the 23-kb molecular weight marker, in contrast to what we observed in our Southern blot analysis (Fig. 1). The reason for this anomaly may be that these large fragments are not easily eluted from the agarose gel under the conditions we are using, unlike in the Southern blotting, where the DNA is first treated with alkali to generate the small fragments required for efficient capillary transfer.”
224. Although the authors state they took “extreme care” to avoid sample contamination from maternal cells, Prof Thierry’s opinion was that they had been significantly contaminated by genomic DNA from maternal cell lysis. While Li 2004 used a double centrifugation step, there were other sources of such contamination, and knowledge of how to avoid such contamination had advanced since 2003/4.
225. If Prof Thierry is correct as to the proportion of DNA over 23 kb in Figure 1, this would mean that the data in Tables 1 and 2 of Li 2004 are likely to significantly underrepresent the proportion of DNA over 23kb (and hence over 500bp) that was present in the samples.
226. Prof Thierry estimated the proportion of fragments that are in the band of about 23 kb to be 50% of the total. Prof Lovett pointed out that this failed to take into account the fact that the probe was for a repetitive sequence, which means that band intensity is not proportional to the number of fragments (as it would be for a probe binding to a single-copy genomic sequence). Prof Lovett was not challenged on this evidence. Instead, the point which was put to Prof Lovett in cross-examination was that his view that the proportion was much less than 50% was inconsistent with what the authors of Li had said about there being a “substantial proportion” or “large amounts” of DNA greater than 20 kb. He pointed out that it depended on what Li 2004 meant by a “substantial proportion” or “large amounts”; his estimate was perhaps a few percent.
227. Prof Thierry was forced to concede that Prof Lovett was correct that the multiple copy nature of Alu repeats, to which the Alu probe used in the Southern blot will bind, would hugely magnify the darkness of any given fragment at the top of the blot, meaning that the relative numbers of fragments could not be estimated just by looking at the darkness of the bands. In his defence, he said that the 50% proportion was “just estimation”, but in my judgment it follows that his estimate was unreliable.
228. Prof Thierry then shifted his ground, saying that he had compared genomes, whereas Prof Lovett had compared fragments. Later, he said that this was the correct comparison because circulatory DNA came from cells, although he did not clearly explain why. There is no doubt that Prof Lovett had compared fragments. It was not put to him that he was wrong to do so, still less was Prof Thierry’s point about genomes (whatever it was) put to him. Moreover, as counsel for the Claimants pointed out, this evidence appears to be contrary to the statements made in footnote 21 to Prof Thierry’s first report which indicate that he was comparing fragments.
229. I therefore accept Prof Lovett’s evidence that the proportion of fragments at around 23 kb is much less than 50%.
230. It is common ground that, if the figures given in Tables 1 and 2 of Li 2004 are taken at face value, then the degree of enrichment of foetal DNA achieved by size separating at 500 bp would be 1.9 times for Table 1 and 1.8 times for Table 2. Prof Thierry’s view, based on his 50% estimate for fragments of around 23 kb which are not included in the tables, was that the true figures were around 3.8-fold and 3.5-fold. For the reasons given above, I do not accept the 50% figure. The Claimants accept that Figure 1 shows that there is some DNA of around 23kb present, and as a consequence the degree of enrichment of foetal DNA as a result of size separation would be greater than the data reported in Table 2 of Li 2004 indicate, because the Table does not take account of the DNA at around 23 kb (as Prof Lovett pointed out, Table 1 does not necessarily stand in the same position, because both Figure 1 and Table 2 relate to early pregnancy, whereas Table 1 relates to late pregnancy).
231. Prof Thierry’s opinion was that enrichment of 3.8-fold and 3.5-fold was not credible and that, as discussed above, the most likely explanation for the results was contamination of the samples. In my judgment, however, the evidence does not establish that the degree of enrichment in Li 2004 would be significantly greater than that indicated by the reported data. Prof Thierry may be correct that, notwithstanding the authors’ statement that they took extreme care, there was some contamination which nowadays could be avoided by better methods of sample handling. I am not satisfied that there was a significant degree of contamination, however.
232. In any event, as the Claimants point out, nothing in Li 2004 undermines the proposition that there is a difference between the size distribution of foetal cell-free DNA and the size distribution of maternal cell-free DNA which enables the foetal fraction to be enriched through size separation at 500 bp. The only question is as to the extent of the enrichment that can be achieved.
Cheng 2015
233. Cheng et al , “Noninvasive Prenatal Testing by Nanopore Sequencing of Maternal Plasma DNA: Feasibility”, Clin Chem , 61(10), 1305-1306 (2015) (“Cheng 2015”) reports a study using a nanopore sequencing device called a MinION to sequence cell-free DNA from women pregnant with male foetuses (third trimester), women pregnant with female foetuses (third trimester), non-pregnant women, and men. The maximum length of fragments sequenced was 5776 bp.
234. Cheng 2015 reports 0.06%–0.3% of sequence reads as being greater than 1000 bp (although it does not say in respect of which sample(s) this was – it could have been in respect of non-pregnant women). It does not give any figure for the proportion of reads that were greater than 500 bp, so there is no way of knowing how many it found between 500 and 1000 bp.
235. Prof Lovett’s evidence, relying on a paper by Laver et al , was that the MinION device is heavily biased towards sequencing short fragments. It was suggested to him that the graph in Laver showing this bias does not explain why the proportion of fragments above 1 kb found by Cheng 2015 is lower than the 7.5% of DNA in the 1-1.5 kb band reported by Li 2004. Prof Lovett’s answer was that there was no way of knowing how the MinION used in Cheng 2015 had been tuned for its sequencing runs, since the size distribution it gives can be changed.
236. Counsel for the Claimants submitted that the uncertainties over the data in Cheng 2015, which were generated using a different technology that had barely been explored in the evidence, provided no basis for concluding that there is little or no cell-free DNA in maternal plasma that is greater than 500 bp given that it is known from Chan 2004 that there is such DNA. I accept this submission.
Electropherograms
237. In addition to the three papers considered above, Prof Thierry put forward three figures containing electropherograms in support of his proposition that there is little maternal cell-free DNA above 500 bp (Figures 1-3 in his second report). Counsel for the Defendants did not rely upon this evidence in his closing submissions, but I will deal with it for completeness.
238. Figures 2 and 3 do not relate to samples from pregnant women, so are not probative, given that Chan 2004 showed a significant difference between pregnant and non-pregnant women. That leaves Figure 1, which shows a size profile from a single pregnant woman. This used a technique for which the sizing range was only up to 7 kb, and in any case a single sample hardly provides any basis for challenging the statistically reliable data in Chan.
Conclusion
239. As counsel for the Claimants pointed out, it is difficult to understand why the Defendants say that there is a squeeze between construction and insufficiency such that, unless the claims are limited to enrichment of at least 2.0-fold, they are insufficient. There appears to be no connection between the Defendants’ contention that the results reported in the Patent are exaggerated due to contamination and the issue of construction.
240. Be that as it may, I have concluded that the claims are not restricted to enrichment of at least 2.0-fold, but encompass a lesser degree of enrichment. So construed, I do not accept that the claims are broader than is justified by the technical contribution made by the Patent. The Patent discloses a general principle of technical utility, namely that the foetal cell-free DNA in maternal plasma or serum can be enriched by size separation at 500 bp. The breadth of the claims is commensurate with that technical contribution. It is immaterial that the extent of the enrichment which can be achieved may vary from case to case and may on average be less than 2.0-fold.
241. Counsel for the Defendants submitted that the excessive breadth of the claims was demonstrated by the fact that there would be cases in which there was hardly any cell-free DNA over 500 bp in maternal plasma. Counsel for the Claimants accepted that there could be such cases, although he submitted that on the evidence they would be likely to be very rare, which I agree with. As I have construed the claims, however, they do not cover fractions derived from such plasma samples. I do not accept that the claims would be insufficient even if they were construed as covering fractions derived from such samples. The fact that on very rare occasions the invention is of no practical benefit does not detract from the fact that in the vast majority of cases it is of technical utility.
Discovery as such
242. The Defendants contend that, even if otherwise valid, the claimed inventions are excluded from patentability under section 1(2)(a) of the 1977 Act on the ground that they are discoveries. The Defendants accept, however, that, as the law presently stands, this contention cannot succeed. They ask the Court to make appropriate findings of fact to enable the issue of law to be argued in a higher court if necessary. The Claimants objected to this course, on the ground that it was not possible to judge the relevance and accuracy of the proposed findings of fact without knowing what legal analysis the Defendants sought to advance in reliance upon them. At least absent an explanation of the Defendants’ legal analysis, the proposed facts were contentious.
243. It is sufficient to illustrate the point to refer to the Defendants’ first proposed finding of fact, which is in the following terms:
“maternal blood and its contents are naturally occurring products”.
244. Counsel for the Claimants accepted that maternal blood was a naturally occurring product, but disputed that its “contents” were necessarily naturally occurring products. This would depend on which contents one was referring to, and for what purpose one was asking the question. For example, a sample of plasma was an artificially created product even though it derived from a naturally occurring one.
245. I agree with the Claimants that it is unsatisfactory for the Court to be asked to make findings of fact in a legal vacuum. As the example I have given illustrates, there is a real danger of the Court being led into making findings which are freighted with legal value judgments. Moreover, I was not referred to any specific evidence which was relied upon as establishing the facts propounded – counsel for the Defendants simply asserted that the proposed findings were made out on the evidence as a whole. Accordingly, I decline to make the findings requested.
Infringement
246. The Harmony Test includes a size-separation step as a result of which the extracellular DNA in plasma samples substantially consists of DNA consisting of 500 base pairs or less. The only issue argued by the Defendants on infringement was based on the 2.0-fold enrichment construction which I have dealt with in paragraphs 112-113 above. It was common ground that the size-separation results, on average, in enrichment of the proportion of foetal cell-free DNA present in the sample. The average extent of the enrichment is agreed, but confidential. There is no need for me to set out the precise figure in this judgment, but it is less than 2.0. The Defendants contend that a substantial part of the enrichment is due to the removal of contaminating maternal DNA released by cell lysis. It is common ground that that some of the enrichment is due to this. Prof Lovett gave unchallenged evidence that about 1/6 of the enrichment was attributable to this effect having regard to the method of centrifugation employed. Prof Thierry suggested that an additional, albeit secondary, contributing factor was agitation of the samples, but it was not shown that this made a greater difference than Prof Lovett estimated. In my judgment the fact that a minor proportion of the enrichment is due to removal of contaminating maternal DNA is irrelevant to the issue of infringement. As I have construed claim 1, TDL has infringed it.
Summary of principal conclusions
247. For the reasons given above, I conclude that:
i) the claims are not obvious in the light of Ikeda;
ii) the claims are not insufficient; and
iii) TDL has infringed at least claim 1.
