- This trial was principally concerned with EP (UK) 3 575 792 ('EP792' or 'the Patent'). The action was commenced by the Claimants ('Sandoz') seeking revocation of the Patent and certain declaratory relief in view of the existence of further divisional(s) still in the process of prosecution. The Defendant ('Biogen') counterclaimed for infringement, as the registered proprietor of the Patent. Since both infringement and validity remained in issue, Biogen opened the trial.
- The Patent is entitled 'Method of assessing risk of PML'. The parties agreed the priority date was 20th April 2012 (the 'Priority Date'). PML stands for Progressive Multifocal Leukoencephalopathy, which is a rare but very serious neurological condition with a high fatality rate.
- Biogen has, since 2006, marketed a treatment for relapsing-remitting multiple sclerosis ('RRMS') called Tysabri, in which the active ingredient is the monoclonal antibody natalizumab. Natalizumab was protected by a patent and by an SPC which expired in July 2020. Sandoz has been developing a biosimilar natalizumab product for treatment of RRMS called Tyruko, for which marketing authorisation was granted by the MHRA on 9 October 2023. Biogen does not assert that it has any UK rights which would be infringed by Sandoz's Tyruko product.
- Natalizumab is generally an effective and well tolerated treatment for RRMS. However, in 2005, during the course of phase III trials, three cases of PML were detected in patients being treated with natalizumab. PML was known to be caused by the John Cunningham virus ('JCV'). JCV infection is widespread in the population and is generally benign, but occasionally the virus can reactivate and lead to PML, particularly in individuals with suppressed immune systems. By the priority date it was well established that treatment with natalizumab could in some individuals lead to reactivation of JCV and the development of PML, and that prior infection with JCV was a pre-requisite for this occurring. It was also well established that MS patients who tested positive for anti-JCV antibodies were at a higher risk of developing PML than patients who tested negative for anti-JCV antibodies.
- By 2012 it was understood that, in addition to JCV infection (as measured by the detection of JCV antibodies in a patient's sample), the risk factors associated with developing PML were: (1) whether or not the patient had been on immunosuppressive drugs; and (2) the length of time the patient had been treated with natalizumab. The following estimates of risk had been published in a review article by Kappos et al in 2011:

- As can be seen from these data, a patient who had been treated with natalizumab for up to 4 years, and was JCV antibody negative, had a ≤0.11 in 1,000 chance of developing PML. For a patient who was JCV antibody positive, the incidence was, for the first 2 years of treatment, three-fold higher at 0.35 in 1,000, rising to 2.5 in 1,000, if treatment was for 2-4 years. Patients were counselled in relation to these risks and based on this information would choose whether or not they wanted to be treated, or continue to be treated, with natalizumab. Many patients were faced with the agonising dilemma of having to face recurrence of episodes of RRMS, which were otherwise being kept at bay by natalizumab, balanced against the alarming potential complication of proceeding with natalizumab therapy and developing PML.
- Notwithstanding the issues over construction and validity which I outline below, there is no doubt that the work summarised in the Patent has provided a better method of evaluating that risk of developing PML.
- Biogen has offered and continues to offer clinicians (free of charge) an antibody assay which is said to implement the teaching of the Patent. It is called Stratify JCV DxSelect ("DxSelect") and, for patients and clinicians based in Europe (including the UK), it is carried out by Unilabs in Denmark.
- The teaching and the assay have been rapidly adopted by clinicians. In assessing the risk of developing PML, clinicians no longer just consider the binary factor of whether a patient is positive or negative for JCV antibodies but monitor antibody levels. If antibody titres are above an index value of 1.5 then patients are considered at high risk of developing PML. Even today, in assessing the risk of PML, clinicians use this factor. The current PML Risk Estimates Algorithm produced by Biogen and published in the PIMG (Physician Information and Management Guidelines) for Tysabri is shown below:
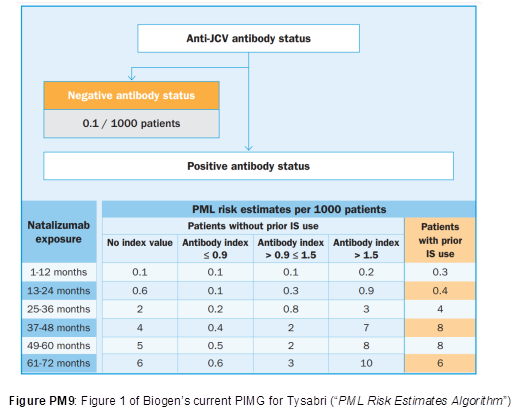
- Sandoz wished to provide an assay for measuring JCV antibody levels, for the same purpose of assessing risk of developing PML, to accompany the launch of its biosimilar natalizumab (Tyruko). In conjunction with their Tyruko product, Sandoz have been developing their own assay for detecting anti-JCV antibodies. Clinicians will be able to submit patient serum samples for analysis by that assay. The assay is known as the ImmunoWELL JCV IgG Test, but it was referred to at the trial simply as the Sandoz Assay. The Sandoz Assay will be conducted outside the UK by a third party.
- These proceedings were issued in September 2021 to clear the way for the launch of the Tyruko product. Initially these proceedings related to EP(UK) 2 676 967, which had claims to natalizumab for use in a method of treating RRMS which involved certain steps dependent on the result of an anti-JCV antibody assay. Sandoz sought revocation of the EP967 patent and Biogen counterclaimed for infringement. The trial was originally fixed for November 2022. However, the EPO TBA fixed the hearing of Biogen's appeal against the revocation of the EP967 patent by the Opposition Division for 7 December 2022, so the trial was vacated and refixed for February 2023 on certain undertakings from Biogen. In the event, the TBA revoked EP967. As a result of the undertakings given by Biogen, no UK rights arising from the EP967 patent family can now be asserted against Sandoz.
- Meanwhile, on 30 November 2022, the Patent was granted. Biogen indicated its intention to enforce the Patent against Sandoz and first provided draft pleadings on 6 December 2022 to introduce the Patent into the current proceedings. On 21 February 2023 Marcus Smith J gave Sandoz permission to amend these proceedings to seek revocation of the Patent, gave Biogen permission to counterclaim for infringement, and fixed an expedited trial for November 2023. I understand that the trial was expedited because it was in the interests of Sandoz and the general public for the question of whether Sandoz was free to launch the Tyruko product and the Sandoz Assay to be determined as soon as possible.
- The Patent explains that it seeks to delineate high and low PML risk groups amongst anti-JCV antibody positive patients based on their anti-JCV antibody titers (using the US spelling as in the Patent) as measured in an anti-JCV antibody ELISA and expressed as index values. Claim 1 of the Patent is as follows:
"A method of evaluating a patient's risk of developing Progressive Multifocal Leukoencephalopathy (PML), the method comprising:
(i) determining, in a serum or plasma sample of the patient, an anti-JC Virus (JCV) antibody titer, wherein the anti-JCV antibody titer is determined by an ELISA assay comprising the following steps:
(a) forming a reaction mixture comprising an aliquot of sample and a substrate on which is disposed Highly Purified Viral-Like Particles (HPVLPs), and
(b) detecting the level of anti-JCV antibody bound to said substrate on which is disposed HPVLPs;
wherein the anti-JCV antibody titer is expressed as an index value, wherein the index value is determined by normalizing an optical density (OD) value of the sample to a cut-off calibrator adjusted to have an nOD of 1, and a positive control is adjusted to have an nOD of 1.3; wherein the cut-off calibrator and positive control comprise a mixture of serum positive for anti-JCV antibodies and serum negative for anti-JCV antibodies, and wherein a negative control comprises anti-JCV antibody negative serum and has an nOD of 0.1; and
(ii) determining the patient to be at high risk of developing PML if the anti-JCV antibody index value is determined to be > 1.5."
- Biogen has applied unconditionally to amend the claims of the Patent so as to delete references to % inhibition. Thus granted claim 2 is deleted and consequential amendments are made to dependent claims. Although Biogen asserted that proposed amended claims 2, 3, 7 and 8 are independently valid, the debate at trial revolved around claim 1 and claim 8 (as proposed to be amended).
- Sandoz pursued a number of grounds of invalidity. Those live at trial were:
i) Classical insufficiency;
ii) Breadth of claim (i.e. Biogen) insufficiency;
iii) Uncertainty insufficiency;
iv) Lack of technical contribution;
v) Lack of inventive step over two pieces of prior art, both publications from Biogen:
a) A 2010 paper in Annals of Neurology by Gorelik et al. called "Anti-JCV Virus Antibodies: Implications for PML Risk Stratification" ('Gorelik');
b) PCT application WO 2011/085369, published in July 2011 ('WO369');
vi) Added Matter; and
vii) Two excluded subject-matter objections.
- On infringement, the issues fall into two categories:
i) First, there is a territorial issue which arises from the fact that the Sandoz Assay will be carried out outside the UK (albeit in an EU Member State). Sandoz contend that the claimed method will not be used in the UK nor offered for use in the UK;
ii) Second, two technical issues arise, which appeared largely to turn on construction. Biogen say that Sandoz infringe either on a normal interpretation of claim 1 or alternatively on the basis of equivalence.
- This outline of the issues conceals the usual suspect: the crucial issue in this case is how to interpret claim 1.
- Finally, Sandoz seek certain declaratory relief which they say is necessary to achieve commercial certainty regardless of any further divisional patents that may emerge. This is characterised as akin to Arrow relief, but Biogen says it is not. Biogen resists the grant of the declaratory relief sought on the basis that it would not serve a useful purpose and/or would not be a proper exercise of the court's discretion.
- Sandoz relied on three witnesses of fact: Mr Gavin Will, Dr Marc-André Frese and Dr Michael Andersen, but Biogen only chose to cross-examine Dr Andersen.
- Mr Will is the Business Unit Head for Bio and Specialty at the Second Claimant. In his third and fourth witness statements (for trial), he explained:
i) some of the background to Sandoz's marketing authorisation for Tyruko and to the development of the Sandoz Assay;
ii) the way in which clinicians in the UK will be able to obtain access to the Sandoz Assay;
iii) Sandoz's expectation as to how Tyruko and the Sandoz Assay will be purchased and used by the NHS; and
iv) the impact of any ongoing uncertainty over Sandoz's ability to provide Tyruko and the Sandoz Assay.
- Dr Frese is a principal consultant at, and co-owner of, Frese Biopharma Consulting and was previously director of regulatory affairs at Polpharma (the Fourth Claimant) and, before that, manager of regulatory affairs at bioeq GmbH. In those roles, he was responsible for regulatory oversight of the projects leading to the development of Tyruko and the Sandoz Assay. He explained the regulatory background to the approval of Tyruko and the development of the Sandoz Assay. In particular, he explained:
i) how it became apparent that bioeq / Polpharma should aim to develop an assay that produced results which correlated with those of STRATIFY JCV DxSelect;
ii) the fact that the company originally appointed to do so was unable to do so and was replaced by GenBio in 2019;
iii) the approach that bioeq / Polpharma adopted to examining that correlation, namely to compare (i) the results which had been obtained by Quest Diagnostics (one of Biogen's partner laboratories) using STRATIFY JCV DxSelect on serum samples taken during the clinical trials of Tyruko with (ii) the results later obtained with the Sandoz Assay developed by GenBio on those same serum samples; and
iv) the EMA's response to the results so obtained.
- Dr Andersen is the Quality Assurance Manager at GenBio and was personally involved in the development of the Sandoz Assay from the initiation of the project. He explained how the Sandoz Assay was developed. Sandoz point out that they had access to serum samples which had been tested using the STRATIFY JCV DxSelect assay, together with the results of such testing. That meant that GenBio could compare its results with those which had been obtained using the STRATIFY JCV DxSelect assay and adjust the design of its assay accordingly. The Skilled Team trying to implement the claimed invention of the Patent would not have had that advantage, but even so it took GenBio about 18 months to produce the Sandoz Assay.
- The parties had permission to call up to three expert witnesses: one expert in the field of human polyomaviruses, one expert in the development of immunoassays and one clinical expert in the field of neurology.
Sandoz's experts
- Mr Simon ('Sam') Scrimshaw has over 25 years' experience developing immunoassays and is the Immunoassay Development Director at Fleet Bioprocessing. At the priority date he was Head of Immunodiagnostic Development at Lab 21, where he was the scientific lead on the development of commercial immunodiagnostic assays, including ELISAs. Mr Scrimshaw has been involved in the design or development of immunoassays for detecting antigens specific to, amongst others, syphilis, malaria, cytomegalovirus and Covid-19. His evidence concerned, in the main, immunoassay aspects of the common general knowledge, aspects of the prior art and the Patent concerning assays, insufficiency and infringement.
- Professor Thomas Berger is a Professor of Neurology, the Chair of the Department of Neurology, and the Chair of the Comprehensive Centre for Clinical Neurosciences and Mental Health at the Medical University of Vienna, Austria. He has been board certified in neurology and psychiatry since 1998 and his research focuses on neuroimmunology including MS. At the priority date, he was the Head of the Neuroimmunology Research Unit and the Neuroimmunology and Multiple Sclerosis Clinic at the Medical University of Innsbruck. His evidence mainly addressed clinical aspects of the common general knowledge, consideration of the cited prior art and the Patent, and obviousness.
- Sandoz served a report from Dr Aisling Dugan who is a Senior Lecturer in Biology, Molecular Microbiology and Immunology at Brown University in the USA, with over 20 years' experience in microbiology research and teaching, including 6 years researching polyomaviruses (including JCV). Her report chiefly addressed virology aspects of the common general knowledge. However, Sandoz chose not to call Dr Dugan for cross-examination, so her report is not in evidence. Biogen contended I should discard her report in its entirety, but Sandoz submitted that in so far as paragraphs from her evidence were adopted or agreed to by other witnesses, the text in those paragraphs becomes the evidence of those other witnesses, just as in the case of any other document, such as a scientific paper. I agree.
Biogen's Experts
- Mr Dene Baldwin is an independent assay development consultant. For most of his career (1994–2014) he worked for Cozart (or successor companies) with a particular focus on assays for drugs of abuse. His evidence primarily concerned immunoassay aspects of the common general knowledge, immunoassay aspects of the prior art and the Patent, insufficiency and infringement.
- Dr Paul Molyneux is the Interim Executive Medical Director at the West Suffolk Hospital Foundation Trust in which he holds executive responsibility for the medical workforce, medical training, research and development, medical revalidation and shares responsibility for patient safety. Between 2003-2021, he was a Consultant Neurologist at the Trust and Addenbrookes where he regularly saw MS patients. His evidence was chiefly concerned with clinical aspects of the common general knowledge, clinical aspects of the prior art and the Patent, and inventive step.
- Professor Polly Roy is, and has been since 2001, Professor of Virology in the Department of Infection Biology at the London School of Hygiene and Tropical Medicine. Her evidence principally concerned the production of virus-like particles (VLPs).
- To the extent that there were criticisms of the experts, I address these in context below. Generally, however, I am very grateful to all the experts for their evidence and assistance in what I found to be a technologically and conceptually complex case. I should also express my gratitude to all the teams of lawyers involved. Although Dr Tappin KC and Dr Turner KC undertook most of the heavy lifting on the counsel side, I was pleased to see Ms Pickard and Mr Alkin being given the opportunity to make submissions on certain topics, including infringement, added matter, excluded subject matter and the declaratory relief.
- Although initially there appeared to be a slight dispute between the parties as to the composition of the Skilled Team, this appeared to resolve itself in the opening skeletons. The Patent is directed to a team comprising a clinician with an interest in treatments for MS ("the Skilled Neurologist"), a virologist with knowledge of JCV ("the Skilled Virologist") and an immunoassay development scientist ("the Skilled IDS", referred to by Biogen as the Skilled Assay Expert). The slight dispute concerned Biogen's contention that the Skilled Team would not necessarily include a virologist with knowledge of JCV, but Mr Baldwin and Dr Molyneux attributed to the Skilled Neurologist and the Skilled IDS a degree of knowledge of virology and JCV, so there was almost no practical difference. Generally, I will refer to the Skilled Team, with two exceptions: first, where the issue specifically engages a particular member of this notional Team and second, when considering what the Patent meant to or taught the Team - in that context I prefer to refer to the Skilled Reader of the Patent, but it is the same Team. Naturally, I have kept in mind the standard attributes of this notional Team.
- The parties cooperated to produce a statement of agreed CGK. By the time of closing argument, there did not appear to be any disputes over CGK or none of any significance. Since the content of this section is essential for understanding the teaching in the Patent, I set it out here rather than in an Annex. This is based on the agreed statement with a few edits of my own.
- The Central Nervous System ("CNS") is made up of the brain and the spinal cord, which are immersed in a fluid called cerebrospinal fluid ("CSF"). The CSF offers a layer of protection to the CNS and also creates an environment in which nerve fibres can transmit signals effectively. The CNS is connected to the rest of the body via the nerves of the peripheral nervous system.
- In a healthy CNS, nerve cells (known as neurones) transmit signals across the CNS by conducting electrical signals along their fibres (known as axons). Neurones are generally made of up of: (1) dendrites, which receive electrical signals from other cells; (2) the cell body (also called the 'soma'), which integrates incoming information; (3) the axon, which carries electrical signals from the cell body to the axon terminal; and (4) the axon terminal which appears at the synapse (the site of intercellular communication where the electrochemical signal is transmitted from one neurone to another).
- The axon of each nerve cell is surrounded by a layer of myelin, a lipid-rich material that is produced by cells of the CNS called oligodendrocytes (in the peripheral nervous system, myelin is produced by Schwann cells instead). The insulating layer of myelin allows electrical signals to be conducted quickly along the axon, assisted by small interruptions called nodes of Ranvier that allow nerve impulses to jump from node to node.
- The endothelial cells of the blood vessels of the CNS tightly regulate the movement of ions, molecules, and cells between the blood and the brain. This separates the brain from the vascular system and is referred to as the blood-brain barrier. In a healthy individual, the blood-brain barrier restricts the passage of elements of the immune system into the CNS, including antibodies, immune cells and signalling molecules. This precise control of movement of molecules and cells between the blood and the brain allows for proper neuronal function and also protects the neural tissue from toxins and pathogens and reduces the risk of an inflammatory immune response.
- Multiple Sclerosis ("MS") is a chronic inflammatory autoimmune disease of the CNS which can manifest in motor and cognitive neurological symptoms of both an episodic and progressive nature. The cause of MS remains unknown but in 2012 it was understood to be triggered by a combination of genetic and environmental factors.
- MS is more common in the female population than the male population and it is typically diagnosed between the ages of 20 and 40. MS threatens an apparently infinite variety of debilitating symptoms and has an unpredictable course. People with MS may encounter a variety of neurological symptoms, depending on the location of the damage in the brain and CNS. Symptoms are highly patient-specific, but commonly include fatigue and weakness, reduced balance, muscle spasms, impaired motor and bladder / bowel function, sensory loss, changes in vision, cognitive impairment and problems with speech and swallowing. Depending on the severity of the disease, MS can lead to severe disability in a short space of time, particularly without treatment. Having MS has a major impact on lifestyle, employment, family life and independence and many people with MS also experience depression and anxiety. In more severe disease, patients become bedridden and may lose the ability to communicate effectively or to eat or swallow. MS patients have a reduced life expectancy of approximately 5-10 years, with an average lifespan of about 30-40 years from diagnosis, although the disease is rarely cited as the primary cause of death.
- In MS, the patient's immune response malfunctions such that it targets the body's own CNS. This auto-immune response results in inflammation which, over time, leads to degradation of myelin (see Figure 1 below), causing disruption of neuronal transmission and giving rise to the range of symptoms described above.
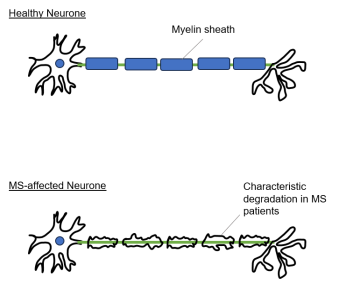
Figure 1: Comparison of neurones in a healthy individual (top) and an MS patient (bottom).
- The damage appears to be caused primarily by T cells crossing the blood-brain barrier into the CNS, where they release chemicals that lead to inflammation and damage to myelin, nerve fibres and oligodendrocytes (the cells that produce myelin in the CNS). B cells and antibodies are also involved in causing damage following activation by T cells. Over time, the damage leads to scarring and hardening of the tissue, hence the name 'multiple sclerosis'.
- Although the patterns of disease and symptoms observed in MS patients are highly variable, MS is classified into clinically important categories in order to predict disease progression and, crucially, treatment options, as most MS therapies are approved to treat only certain types of MS. MS cases are most commonly categorised into:
i) relapsing-remitting MS (RRMS) - the most common form of MS, affecting at least 70-80% of patients upon initial onset of the disease. It is marked by "flare ups" of new or recurrent neurological symptoms developing over days to weeks (relapses or exacerbations) followed by periods of remission, when symptoms improve or disappear. A relapse is defined as new or worsening symptoms lasting for at least 24 hours, although a relapse may persist for days. The frequency and duration of relapses will vary between patients, as will the severity of the relapses.
ii) primary progressive MS (PPMS) - affects approximately 10% of MS patients and is characterised by steady progression of disability from disease onset. Symptoms continue to worsen gradually from the beginning. There are no relapses or remissions, but there may be occasional plateaus.
iii) secondary progressive MS (SPMS) - will eventually develop in the majority of patients with relapsing–remitting disease. SPMS is characterised by an initial period of relapses (often initially categorised as RRMS) followed by a gradual reduction in relapses and remissions while the disease progresses; and
iv) progressive relapsing MS (PRMS) - a rare form, affecting fewer than 5% of patients. It is progressive from the start, with intermittent flare-ups of worsening symptoms along the way and no periods of remission.
- PPMS, SPMS and PRMS are much less common than RRMS and, therefore, less commonly targeted by MS treatments.
- In 2012 it was understood that periods of inflammatory activity correlated with relapse events. Patients experiencing a progressive disease would experience less inflammation but increasing permanent neurodegeneration as a result of damage to tissue in the CNS.
- In 2012, MS was diagnosed by reference to the widely used and well-established McDonald criteria. Diagnosis of MS involved examining the patient's history, conducting a neurological examination, the use of magnetic resonance imaging ("MRI") to observe damage to the white matter of the CNS (referred to as lesions), and testing the patient's CSF for the presence of oligoclonal bands (IgG antibodies, indicating an immune response within the CNS). As part of the patient's neurological examination the clinician would have referred to one of the scales used to assess disability e.g. the Kurtzke Expanded Disability Status Scale ("EDSS") which is a scale from zero (no neurological impairment) to ten (death caused by MS).
- Diagnosis and characterisation of MS would be reached based on evidence of damage to the CNS, i.e. MS lesions occurring in multiple parts of the CNS at different points in time. MRI scanning is also used to monitor progression of the disease.
- A patient's type of MS, the severity and nature of symptoms presented, and also the MRI findings would be taken into account when determining the most appropriate course of treatment.
- At the priority date, there were no treatments to cure MS or reverse the disabling effects that it causes. Treatments for RRMS in 2012 focused on modulation of the immune system to reduce inflammation in the CNS and reduce the number of relapses. Only a small number of licensed treatments were available for treating MS in the UK.
- MS treatments targeted only the disease's symptoms until interferon beta treatments (commonly called interferons), the first disease modifying therapies ("DMTs") for MS, were developed in the 1990s. Interferons, which are cytokine proteins associated with cell signalling during the inflammatory response, treat the underlying disease but are only capable of modestly improving relapse rate and disability progression in RRMS patients, and have an even lesser effect in SPMS patients. The three interferon beta therapies approved for treating MS at the priority date were Avonex, Rebif and Betaferon.
- In the early 2000s, treatment alternatives to interferons became available. Glatiramer acetate (marketed as Copaxone), a random polymeric mixture comprising four of the amino acids present in myelin, was approved in the UK in 2002 and treated RRMS with similar efficacy to the interferons.
- "Off-label" treatment with immunosuppressive medicines were sometimes used to treat MS but by 2012 these were rarely used in the treatment of RRMS in light of modest results and a high risk of severe side effects.
- At the priority date, a new, oral DMT fingolimod (marketed as Gilenya), had recently been approved by the European Medicines Agency ("EMA") for use in severe cases of MS. It was not available in the UK until after its NICE (National Institute for Health and Care Excellence) technical appraisal, which followed shortly after the priority date in 2012 and recommended its use in patients with highly active RRMS.
- Natalizumab (developed and marketed by Biogen under the brand name Tysabri), was approved by the EMA in June 2006 and received its NICE technical appraisal in August 2007. It was the first monoclonal antibody approved specifically for the treatment of MS and was approved for use in patients with "rapidly evolving severe" RRMS (severe cases of RRMS meeting certain criteria) or patients with high disease activity despite treatment with interferon beta, even though the NICE approval only covered the former.
- Natalizumab is a monoclonal antibody that works by blocking the interaction between proteins on the surface of activated T cells (a type of white blood cell or lymphocyte) known as α4β1 integrins or "very late antigen-4" (VLA-4) and the vascular cell adhesion molecule on the endothelial surface (VCAM-1 receptors). α4β1 integrins are involved in the migration of lymphocytes across the blood brain barrier and into the CNS, where they play a critical role in recognising and eliminating foreign pathogens (known as "immunosurveillance"). By inhibiting α4β1 integrin, natalizumab was understood to prevent T cells from entering the CNS and triggering the inflammatory response that causes the characteristic damage to myelin and neurones of MS patients. A diagram illustrating the effect of natalizumab on the ability of a T cell (lymphocyte) to cross the blood vessel's endothelium and enter the CNS (parenchyma) is shown in Figure 2 below.
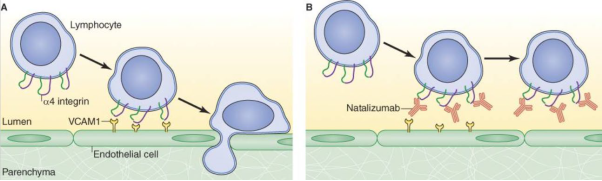
Figure 2: Effect of natalizumab on the ability of a lymphocyte to enter the CNS.
- When developing a new drug or therapy, there are several stages or "phases" of clinical trials that the product must go through in order to demonstrate its safety and efficacy to the relevant regulatory bodies, enabling its approval for use as a medicine. Phase I clinical trials are small studies assessing the safety and pharmacology of a therapy in (usually healthy) humans, following pre-clinical laboratory and animal studies. Phase II clinical trials are expanded human studies testing a larger number of individuals, usually including patients with the disease, which also assesses dosage and mode of administration. Provided the drug succeeds in its initial two phases, Phase III trials will then test the drug on a larger number of patients, aiming to assess clinical efficacy rates and monitor for incidence of any adverse events. Sometimes a fourth round of trials, Phase IV, are initiated after regulatory approval in order to evaluate further the properties of a product in the post-marketing phase.
- Following successful Phase I and II results, two large multi-centre, randomized double-blind placebo-controlled Phase III trials for Tysabri in MS patients were carried out in the US. In one trial, AFFIRM, patients received Tysabri as a monotherapy while in the other, SENTINEL, patients received Tysabri in combination with interferon Avonex.
- The one-year preliminary results arising from the AFFIRM and SENTINEL trials were positive, showing highly significant reductions in clinical relapse rates and detection of MS-induced lesions in MS patients compared to the control groups. In the AFFIRM trial, natalizumab administration resulted in a significant reduction in clinical exacerbations and the annualised relapse rate represented an almost 70% reduction - a substantial improvement over the existing therapies (interferon beta and glatiramer acetate), which reduced the annualised relapse rate by around 30%.
- In June 2004, Biogen filed an application with the FDA for approval of Tysabri for treatment of RRMS and, on the basis of the success demonstrated by the early Phase III results, the FDA designated Tysabri for Accelerated Approval and Priority Review, a mechanism which reduces the time taken for approval of medicines that represent significant improvements in treating serious conditions or that address unmet clinical needs.
- Shortly thereafter, in November 2004 it was announced that the FDA had granted accelerated approval to Tysabri.
- On 28 February 2005, soon after Tysabri launched in the US, Biogen announced voluntary suspension of its commercial distribution of the product and suspension of all ongoing clinical trials, based on reports of two serious adverse events identified in the Phase III patient population. These events included one confirmed fatal case and one suspected (non-fatal) case of the extremely rare neurological disease PML, in MS patients who were receiving Tysabri in combination with Avonex on the SENTINEL trial. Shortly afterwards, a third case of PML was identified following re-examination of cause of death in a previously treated patient. The initial case reports were published in the New England Journal of Medicine alongside a review of these cases.
- In June 2006, the EMA approved Tysabri for marketing in Europe for the first time, on the basis that Biogen would introduce various measures to improve the safety of the drug. These measures mirrored the approach taken by the FDA, which had reauthorised Tysabri earlier in the same month. Those measures included restricting use of Tysabri to RRMS patients as a monotherapy, adding information regarding PML risk and recommending additional MRI monitoring to product labelling, and establishing a risk management plan. Biogen also established mandatory restricted distribution programs involving regular monitoring by certified healthcare providers, and a post-marketing safety study (known as TYGRIS) to assess incidence of PML and other serious adverse events in patients receiving Tysabri.
- By 2012, Tysabri had been in use for a number of years despite the risk of PML. According to a 2011 review in Lancet Neurology by Kappos et al. ("Kappos") by 31 March 2011 around 83,300 patients worldwide had been treated with natalizumab, although the Skilled Neurologist would appreciate that the number would have gone up by April 2012. Cases of PML in natalizumab patients had increased in the years since it re-entered the US market and was first marketed in Europe. According to Kappos, the number of cases had reached over 133 by 1 June 2011.
- The high uptake of Tysabri following its launch was due to its superior efficacy by comparison to other available treatments. By the priority date, MS neurologists were able to advise patients on their estimated PML risk according to the established risk factors (addressed in section F below) and supportive paraclinical tests, which neurologists used when discussing the benefits and known risks of treatment. Further, many patients suffering the debilitating effects of MS were willing to run the small risk of developing PML (despite its serious consequences) to obtain the benefits that the treatment might have on their condition (with its risk of severe disability and often life-limiting effects). This was a benefit/risk assessment which the patient would make in conjunction with the clinician.
- PML is a rare but very serious brain disorder, characterised by the destruction of oligodendrocytes, resulting in lesions in the white matter of the CNS and severe neurological impairment. An individual who develops PML is at high risk of a fatal outcome within 6 months of diagnosis. There is no effective treatment for PML and those that do survive are generally left with debilitating neurological impairment.
- PML can produce a broad range of motor and cognitive symptoms, depending on the location of damage to the CNS, but the symptoms are typically more profound and relentlessly progressive than those observed in MS patients.
- Historically, PML had been associated with severely immunocompromised patients, most commonly being observed in cases of untreated AIDS, severe cases of leukaemia and lymphoma, and recipients of organ transplants receiving immunosuppressant drugs to prevent organ rejection. PML was only associated with MS after the incidences that occurred during the clinical trials for Tysabri.
- PML is caused by JCV, a virus with which a large proportion of the human population is infected. Most healthy individuals present no noticeable symptoms of having been infected. Infection predominantly occurs during childhood. Antibodies arise during initial infection and typically remain throughout the lifetime of the infected individual.
- JCV is usually present in the body as a persistent infection, meaning that it replicates slowly without causing any symptoms.
- JCV is a virus in the family of small non-enveloped viruses called polyomaviruses, of which it is among the most well-known along with BK virus (BKV). Human polyomaviruses are double-stranded DNA viruses, and their DNA is packaged as a superhelix (in which a helix is itself coiled into a helix) with cellular proteins called histones.
- The human polyomaviruses are relatively small in terms of genome and capsid size. The viral capsid is comprised of three structural viral proteins: VP1, VP2 and VP3. On the surface of the JCV capsid, five VP1 particles congregate into pentameric subunits. 72 pentamers make up the surface of JCV, which forms an icosahedral (or broadly spherical) 3D shape. VP2 and VP3 are also constituents of the viral capsid. For each VP1 pentamer, one VP2 or VP3 protein sits within a small pore at the centre. VP1 makes up the major portion of the viral capsid (hence it is referred to as the major capsid protein) and, being exposed on the surface of the capsid, is the major protein involved in viral attachment to cells. VP2 and VP3 are referred to as minor capsid proteins. The structure of JCV is illustrated in Figure 3.

Figure 3: Structure of JCV.
- JCV remains in peripheral tissues including, typically, the urinary system and particularly the kidneys.
- JCV DNA is occasionally shed in urine of those infected with JCV during asymptomatic reactivation events in healthy individuals. It was not known what caused these shedding events, although they were more commonly identified in pregnant women.
- The presence of JCV DNA in the blood or the urine indicated that the person had been exposed to JCV, but the absence of JCV DNA from either blood or urine did not indicate whether the patient had been infected.
- In 2012, it was common to test for prior exposure to JCV by testing for the presence of antibodies to JCV in serum.
- In 2012, it was still not well understood what caused JCV to reactivate and lead to PML in some immunocompromised individuals but not others. Given how rare the condition was, even in severely immunosuppressed patients, immunosuppression alone did not appear to account for disease onset. Further, it was not known what the time period was between JCV reactivation and disease onset in those patients that went on to develop PML. In addition to the rarity of PML, another factor that made it difficult to study the mechanisms of reactivation and disease development was the lack of an animal model for the development of PML.
- It was generally considered that JCV was present in the brain and cerebrospinal fluid (CSF) of individuals who developed PML. In 2012, a diagnosis of PML was based on neurological examination and MRI scans and then confirmed by the detection of JCV DNA in the CSF or brain tissue by PCR (polymerase chain reaction).
- By 2012 the risk factors that contributed to PML development had been further elucidated and it was clear that duration of treatment with natalizumab (in particular treatment beyond 2 years), prior use of immunosuppressants (including chemotherapy) and the presence of anti-JCV antibodies were all risk factors for PML.
- Whilst the absolute risk of PML was low, it was known that a patient who was anti-JCV antibody negative was at a much lower risk of developing PML than one who was anti-JCV antibody positive.
- The SmPC for Tysabri specified that:
"Anti-JCV antibody status identifies different levels of risk for PML in TYSABRI treated patients. Patients who are anti-JCV antibody positive are at an increased risk of developing PML compared to patients who are anti-JCV antibody negative. Patients who have all three risk factors for PML (i.e., have received more than 2 years of TYSABRI therapy, and have received prior immunosuppressant therapy and are anti-JCV antibody positive) have the highest risk of PML at approximately 9 in 1,000 patients treated. Patients should be informed about this increased risk for developing PML before continuation of treatment after 2 years. For risk stratification prior or during the treatment with TYSABRI anti-JCV antibody testing may provide supportive information."
- A risk algorithm, setting out the incidence of PML per 1000 patients based on the three established risk factors was set out in the Physician Information and Management Guidelines (PIMG) for Tysabri. This is shown in Figure 4 below, although it may be noted that another version of this had been published in Kappos.
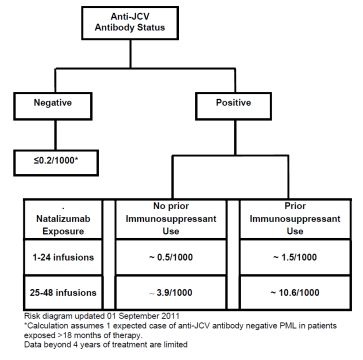
Figure 4: PML risk algorithm from PIMG for Tysabri.
- At the priority date, the Skilled Neurologist would refer to the figures for risk of developing PML provided by the risk algorithm published by Biogen. Biogen regularly updated clinicians with the latest guidance on mitigating PML risk (including the latest risk stratification algorithm) in patients who were being considered for or receiving Tysabri and were in regular dialogue with clinicians providing the latest figures for incidence of PML.
- In 2012, prior to prescribing Tysabri or during a treatment review, patients would be counselled about the risk of PML and advised as to their personal risk factors.
- To determine a patient's JCV antibody status, it was common practice in 2012 to test for anti-JCV antibodies prior to and during treatment with Tysabri via serology testing, i.e. testing for the presence of anti-JCV antibodies in the blood serum.
- Biogen began offering an anti-JCV antibody assay called STRATIFY JCV to clinicians in the UK and Europe in the first half of 2011. The assay was specifically for use in the context of risk assessment prior to and during Tysabri treatment and could determine whether a patient was positive or negative for anti-JCV antibodies. As part of Biogen's testing service, which was offered free of charge, patient blood samples were sent to a third-party partner laboratory in Denmark (namely Unilabs) for testing, and the results of the STRATIFY JCV test were reported to clinicians as being either anti-JCV antibody positive or anti-JCV antibody negative. By 2012, clinicians would routinely send serum samples of RRMS patients to be tested for serum anti-JCV antibodies prior to administering Tysabri.
- While the STRATIFY JCV assay provided clinicians with a binary result, MS neurologists were aware that it was a two-step ELISA (enzyme-linked immunosorbent assay), and that it had been developed and validated specifically for use in patients receiving or considering treatment with Tysabri.
- Those patients who tested positive for anti-JCV antibodies would be viewed as being at a higher risk of developing PML than those who tested negative. Those who tested negative for antibodies to JCV most likely were not infected by JCV and so could be administered Tysabri more safely, although the risk of PML was not eliminated entirely due to the risk of later infection with JCV (and thus seroconversion from negative to positive anti-JCV antibody status) or the possibility of a false negative result from the antibody test (a small possibility in a clinically validated assay). Accordingly, seronegative patients would be retested for anti-JCV antibodies at regular intervals (generally every 6 months) after treatment began.
- Prior to prescribing Tysabri or during a treatment review, patients would be counselled about the risk of PML and advised as to their personal risk factors. For a patient who was anti-JCV antibody negative, the clinician would explain that the risk of PML was extremely low, unless the patient later tested positive. For an individual who was anti-JCV antibody positive, the clinician would explain that there was an increased risk of PML. They would also consider the patient's other risk factors discussed above.
- A seropositive patient might still be treated with Tysabri, particularly given the limited availability of highly effective treatments for RRMS at the time. Given the relative rarity of PML and the significant benefits of Tysabri treatment, some patients with positive results were willing to take the relatively small risk of PML as Tysabri was likely to be effective in preventing or reducing their MS symptoms. However, following consultation with their clinician, other patients would decide not to receive (or not to continue to receive) Tysabri treatment if they tested positive for anti-JCV antibodies.
- Patients who decided to proceed with treatment would be advised about the signs and symptoms of PML and they would be carefully monitored (a baseline MRI would be conducted and thereafter MRI scans would be taken every 6 months to a year). Patients continuing treatment (as opposed to starting treatment) would particularly be advised about the increased risk of developing PML if they continued treatment beyond two years.
- An assay is an analytical test used to detect a target compound referred to as the analyte. A large array of analytes can be assayed using different techniques, which in a medical / pharmacological context may include, amongst many others:
i) DNA or RNA (e.g. from a particular virus);
ii) antibodies (e.g. to a particular pathogen such as bacteria or virus);
iii) proteins (e.g. hormones such as insulin);
iv) small molecules (e.g. hormones such as testosterone or progesterone);
v) illicit substances (e.g. illegal drugs);
vi) medicines (e.g. determining levels of certain medications to assess pharmacokinetics); and
vii) toxins (e.g. heavy metals).
- The detection of an analyte may represent the end in itself (e.g. detection of a viral gene sequence, for example using PCR to confirm infection), or may allow an inference to be made (e.g. determination of the level of a particular hormone to factor into a clinical diagnosis).
- There were innumerable assay techniques available by the priority date. Many assays relied on antibody-antigen interactions, which then may be referred to more specifically as immunoassays.
- An antibody is a large protein (also referred to as an immunoglobulin), raised by the body as part of its immune response to a pathogen, such as a virus or bacteria. The part of a pathogen that triggers the production of antibodies by B cells is the antigen. Antibodies of the IgG class (described below) are typically drawn as a 'Y', illustrating their structure comprising two longer heavy chains of amino acids and two shorter light chains. The 'stem' and lower part of the 'fork' of the Y shape form the constant region, and the upper part of the 'fork' forms the variable region, as shown in Figure 5.
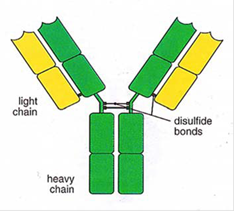
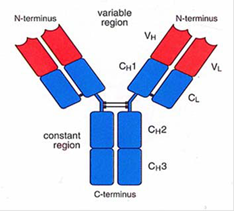
Figure 5: Typical antibody structure showing heavy and light chains (left) and constant and variable regions (right).
- As the name suggests, the variable region differs greatly between antibodies and is primarily (although not solely) responsible for the three-dimensional structure of the critical antigen binding site at the tip of the antibody variable region, known as the paratope. This allows each antibody to target a specific antigen. The paratope on the antigen-binding site of the antibody interacts with a region on the antigen known as the epitope. An antigen is likely to contain a number of different epitopes that may be bound by different antibodies. During binding, complementary interactions occur at the binding site between the amino-acid side chains of the antibody and atoms or groups of atoms on the antigen. The term affinity is used to describe the strength of the interaction between the paratope and the epitope. An antibody with perfect specificity would bind only to one epitope on one antigen, however in reality, specificity is rarely perfect and antibody binding can occur, for example, to non-target antigens with a similar structure at lower affinity, which is referred to as cross-reactivity. Avidity is a measure of the total strength of the antibody-antigen interaction.
- The constant region shows less variation between antibodies but can be used to categorise antibodies into different classes, and the two most important classes are IgG and IgM. When the body first encounters a new pathogen, the primary immune response leads to the creation of IgM antibodies. However, IgM antibodies do not typically persist at appreciable levels for long, and so detecting IgM antibodies is indicative of a recent exposure to a particular pathogen. IgG antibodies are the most abundant class in the blood and form the body's main response for subsequent exposure to a known pathogen, and so detecting IgG antibodies can therefore indicate previous exposure to a given pathogen. The concentration of IgG antibodies varies over time, for example quickly increasing when a repeat exposure occurs, before subsiding again once the immune response has dealt with the exposure, but often remaining at appreciable levels in the blood for years or even decades.
- When describing the body's response to a particular pathogen, the singular 'antibody' is often used as a shorthand, but it is important to note that multiple B cells (key immune white blood cells which produce antibodies, also referred to as B lymphocytes) will each produce different antibodies to a given target. This results in a vast array of antibodies to different antigens which, even for those targeting the same antigen, are subtly different and may attach to a given antigen in slightly different ways, i.e. they will bind to different epitopes on the antigen and with different affinities. These are therefore referred to as polyclonal antibodies or polyclonal serum.
- This is in contrast to monoclonal antibodies produced in a laboratory, in which multiple identical copies of a single antibody are produced, all originating from a single cloned cell line.
- A common class of pathogen that elicits an antibody response is viruses. At the most basic level, a virus comprises its genome (single or double stranded DNA or RNA) surrounded by a protective coat called a capsid. The capsid is formed of repeating protein units which self-assemble to a given structure. Some viruses also have an additional coat called an envelope. Viruses are many and varied and are grouped into families on the basis of shared characteristics, including structure and genome type.
- The characteristics of the capsid, and envelope if present, distinguish different viruses, or classes of virus, from one another.
- Antibodies produced against an infecting virus often target the capsid or envelope of the virus in question, since the surface of the virus is the most visible to the host's immune system.
- Assays for detection of nucleic acids, notably PCR, and various types of immunoassay were routinely in use at the priority date.
- The polymerase chain reaction, or "PCR", was developed in the 1980s and enables minute quantities of DNA in a sample to be amplified by many orders of magnitude to reach a sufficient level to be detectable.
- Developed during the 1940s, haemagglutination inhibition assay ("HAI") was an early immunological test which may be used to detect the presence of anti-viral antibodies. Many common viruses express haemagglutinin glycoproteins on their surface, which bind red blood cell receptors (specifically sialic acid receptors). This interaction causes the clumping (or haemagglutination) of red blood cells, which leads to a distinct change in appearance which can be seen with the naked eye as a red spot in the assay well.
- An immunoassay detects the presence or concentration of a target in a sample by making use of the antibody-antigen interaction. The analyte can be a small molecule such as a protein, or an antibody. An immunoassay relies on an antibody being specific to its antigen. The binding between an antibody and an antigen can be utilised to isolate the analyte from a sample and therefore detect its presence.
Enzyme-linked immunosorbent ass ay (ELISA)
- One type of immunoassay is the ELISA. The first ELISAs were developed in the 1970s and by the priority date had become commonly employed assay techniques, and remain so today.
- ELISAs use an enzyme conjugated to an antibody to produce a signal if an analyte is present. The analyte can be either an antigen or an antibody, and its complementary antibody or antigen is immobilised on a solid surface. If the analyte is present when the sample is passed over the immobilised antibody or antigen, a binding pair is formed in which the analyte is captured. The plate is then washed, leaving any antibody-antigen complexes immobilised on the plate.
- One common form of immunoassay is the indirect ELISA, which is relevant to this case. This method involves the following steps:
i) A viral antigen (the 'capture antigen'), to which the antibody of interest binds, can be adhered directly to the plate (typically a plastic microtitre plate containing 96 wells). The plate is then washed to remove any non-bound antigen.
ii) An inert protein (commonly bovine serum albumin, casein or gelatine) is then added to "block" the plate, and thereby occupy any part of the plastic surface which has not been coated with antigen. The plate is washed to remove unbound material.
iii) The test sample which potentially contains the antibodies of interest (primary antibodies) is added to the plate and, if present, will bind to the adhered antigen. The plate will usually be at room temperature but the temperature of the plate can be controlled in some circumstances. The period of time for which the sample is left to react with the antigen is known as the incubation period. The incubation period will be fixed; incubating for longer may lead to an increase in binding of target antibodies, which could have an effect on the assay output. The plate is again washed.
iv) A secondary reagent antibody (often referred to as the 'detector antibody' or 'conjugate antibody') which binds to the constant regions of human antibodies is then introduced and will bind to the primary antibody on the solid phase that has bound to the antigen on the plate. (The antibody used as the detector antibody will depend on the analyte that it is being used to detect. For example, if the analyte is a human IgG antibody, then the detector antibody will be an anti-human IgG antibody. If the analyte is a viral antigen, then the detector antibody will be an anti-viral antibody. The linking of the detector antibody to an enzyme is known as 'conjugation'. Detector antibodies were readily available in 2012 from commercial suppliers). As with the previous step, there is a fixed incubation period. This antibody will have been conjugated to an enzyme. This produces a colour change when substrate is added and the enzyme catalyses the conversion of the substrate into a coloured product. The plate is again washed.
v) The most common enzyme is horseradish peroxidase (HRP). If HRP is used, a solution containing HRP substrate and hydrogen peroxide is added. The HRP conjugated to the antibody and in turn bound to the antigen catalyses the oxidation of a substrate in the presence of hydrogen peroxide.
vi) Common HRP substrates include 3,3',5,5'-tetramethylbenzidine (TMB), o-phenylenediamine dihydrochloride (OPD) and 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) each of which become chromophoric (i.e. coloured) once oxidised by the HRP and turn blue, amber and green respectively.
vii) To halt the reaction between enzyme and substrate a stop solution is added to the plate, which almost immediately brings the reaction to an end. The identity of the stop solution depends on the enzyme being used. Common stop solutions for HRP include sulphuric acid (H2SO4) and hydrochloric acid (HCl). If TMB is being used as the substrate for HRP, the acidification also causes the colour of the solution in the well to change from blue to yellow.
viii) The optical density (described in further detail below) of the solution in the plate well is then measured using a spectrophotometer.
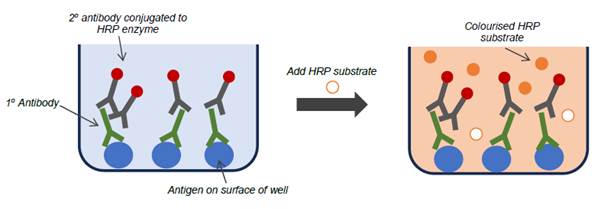 Figure 6: A typical indirect ELISA wherein the target (primary) antibody is present in the test sample, causing a colour change to occur.
Figure 6: A typical indirect ELISA wherein the target (primary) antibody is present in the test sample, causing a colour change to occur.
- The colour change in an ELISA arises from an enzymatic reaction and, as such reactions are catalytic, the enzyme is not consumed when it oxidises the substrate. Accordingly, if left for sufficient time, even a low concentration of enzyme would eventually lead to significant colour development. However, standard practice was to set a fixed incubation period to standardise the time allowed for colour change to develop after which the enzyme would be inactivated by adding a stop solution.
- By the priority date, equipment and many of the reagents for use in developing an ELISA could be readily purchased commercially, for example plates, buffers, HRP-bound antibodies, HRP-substrate etc.
- When testing for the presence of an antibody in a patient sample, competitive ELISAs (also known as inhibition ELISAs) were also commonly used to look for specific antibody binding to the target antigen. In one form of a competitive ELISA, the target antigen is added to patient serum to compete for binding with antibodies that would otherwise bind specifically to the antigen coated on the ELISA plate. In doing so, fewer antibodies will bind to the antigen coated on the ELISA plate, and thus the subsequent colour change in the ELISA is reduced. A reduction in signal therefore indicates the presence of antibodies specific for the target antigen in the patient serum. This format can be useful where it is known that there are issues relating to cross reactivity or non-specific binding of the antibodies in the sample.
- In one form, the patient serum may be incubated with antigen for a period of time prior to being tested in the ELISA, thereby resulting in fewer antibodies available to bind to the antigen coated on the ELISA plate. Alternatively, the free antigen and serum may be combined and then added directly to the well of the ELISA plate, and the free antigen will again compete against the antigen coated on the plate for binding to specific antibodies. Due to kinetic factors, specific antibodies will have a higher binding affinity for free antigen in the serum sample, over the surface-bound antigen of the ELISA. The more of the natural antigen present in the solution phase, the more it will out-compete the antigen coated to the surface of the ELISA plate, such that when the well is washed, and secondary antibodies followed by substrate are introduced, the signal (colour change) is inversely proportional to the amount of specific antibody in the patient sample.
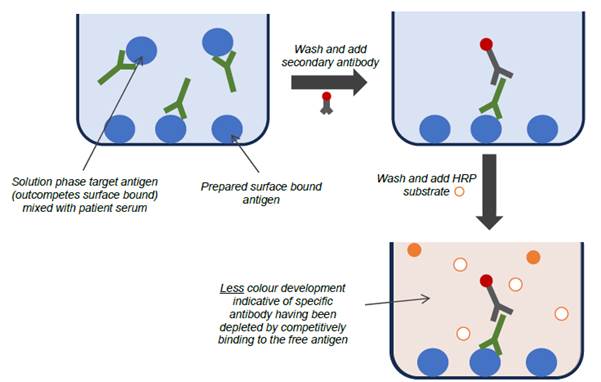
Figure 7: A typical competitive (inhibition) ELISA wherein the target antigen is present in the test sample, reducing colour development.
- As explained at [107] above, since it is standard practice to set a fixed incubation period to allow colour change to develop, it is strictly more accurate to say that a decrease in the rate of colour change in a competitive ELISA is inversely proportional to the analyte concentration, but for brevity this nuance is often left as being assumed to be implicitly understood by those in the field.
- An immunoassay generates a detectable signal to indicate that the antibody-antigen binding has occurred and hence the analyte is present. The signal could be, for example, a colour change, fluorescence, radiation emission or chemiluminescence. ELISAs typically produce a change in colour, and this is detected using a device called a spectrophotometer (or just spectrometer). The intensity of the colour change is related to the amount of analyte present. Often in a standard sandwich or indirect ELISA this is shown as an increase in the intensity of the colour change as the concentration of the analyte increases.
- The output measurement from the spectrometer is the optical density (or "OD"), which is also called the absorbance, of the sample. This represents the ratio of the intensity of light falling on the sample compared to the intensity of light transmitted from it. The OD is measured at a specific wavelength according to the substrate used. For TMB oxidised by HRP, this is at its maximum at 450 nm (following acidification), and so the term OD450 is sometimes seen.
- The OD value produced by an ELISA is logarithmic with base 10. As OD is a ratio, it is a unitless parameter and another way of expressing OD is that it is the negative log of the light transmission through the sample, and hence the two parameters are related as shown in Table 1.
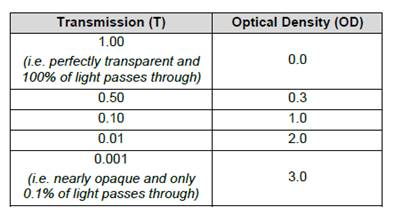
Table 1: Relationship between light transmission and optical density measured by a spectrometer.
- As can be seen in the table, an OD of 1 occurs when 10% of light is transmitted through a sample, whereas by the time the OD reaches 2 only 1% of light is being transmitted. Accordingly, at higher OD values, it becomes increasingly difficult to obtain an accurate and reproducible result, as the intensity of light that needs to be detected becomes extremely low (i.e. there is a smaller absorption range to distinguish differences in analyte level in a sample). Figure 8 below is an image of typical ELISA plates showing the colour development usually seen in blue, amber and green (which were all common options according to the enzyme/substrate system used).

Figure 8: Typical ELISA plates showing colour development in blue (left), amber (middle) and green (right).
- The values of OD that any given spectrophotometer can reliably detect is referred to as the dynamic range (or 'working range'), which is narrower than the OD value operating range of the spectrophotometer i.e. the range of OD values that the equipment can actually measure. Above the top end of the dynamic range, the reliability of the OD values becomes increasingly suspect. For example if a spectrophotometer has an upper limit of its dynamic range of 2.5 OD, a result of 3 OD or 4 OD in effect merely means "greater than 2.5", and so many spectrophotometers simply report a qualitative result (e.g. "high") for results outside their dynamic range. At the lower end of the dynamic range, a blank sample (containing buffer only) typically produces an OD between around 0.05 to 0.1.
- The dynamic range of a given spectrophotometer varies according to the sophistication of the device, but essentially all spectrophotometers at the priority date could reliably detect in at least the 0.1 - 2 / 2.5 OD range. Therefore, when designing an assay that was intended to be run on a variety of devices, the Skilled IDS would seek to ensure that the results would fall into that 0.1 - 2 / 2.5 OD dynamic/working range in so far as possible.
- In most cases an ELISA is intended to test multiple samples in a single run. The most common format of an ELISA is a 96-well plate (commonly made of polystyrene or a similar material) which is an eight by 12 array of plastic wells typically with a working volume of around 0.25 - 0.4 ml for an analytical plate. To account for inherent variation, both in the assay and in the spectrophotometer measurement of OD, it is typical in ELISAs to run each well in at least duplicate and use the mean OD value. For critical components, triplicate or quadruplicate wells may even be used.
- Most ELISAs will have both a positive control and negative control. The positive control will be produced using a sample positive for the target antibodies. The negative control will generally be produced using a sample negative for the target antibodies. The positive and negative control enable the operator to know that the assay is working.
- As noted above, the OD measurements in ELISAs are prone to variability as the assay is susceptible to changes in conditions - for example, a warmer room may increase colour change (i.e. increase OD) as reactions proceed faster at higher temperatures, or an ELISA performed with the same reagents a week apart may see a reduced OD measurement (e.g. as reagents degrade). Therefore, to correct for these factors and allow comparison across different plates, it is typical to normalise the OD ("nOD"), which means report the result of the assay as the OD of the sample relative to a calibrator rather than any absolute OD value.
- ELISAs could be set up to provide results with differing degrees of information, in particular:
i) Qualitative - providing a simple binary result (e.g. positive or negative)
ii) Semi-quantitative - providing a more graduated result (e.g. low, medium, high)
iii) Quantitative - providing a numerical result (e.g. a mg/ml concentration value)
- The nature of the result sought will influence the set up of an ELISA.
- One way ELISAs are used in a qualitative manner is to determine the presence or absence of an analyte. In that case a defined cut-off threshold may be determined at a particular signal output, below which the analyte is deemed to be absent.
- Alternatively, an ELISA may be used to determine whether a sample is above or below a clinically relevant threshold, rather than to determine whether an analyte is present or absent. The clinically relevant threshold is determined in clinical studies using a particular ELISA protocol and calibrator/standards.
- The ELISA could be set up using a calibrator that corresponds with the cut-off threshold that has been determined. In that case, the OD of the unknown sample can be compared to that of the calibrator to see whether it is higher or lower.
- Key terms used at the priority date in the analysis of assay performance are set out below.
Sensitivity – in the clinical context, clinical sensitivity of the assay is a measure of the frequency with which an assay correctly identifies a sample as positive for a particular disease or state. Sensitivity is calculated as:
Sensitivity = True Positives / (True Positives + False Negatives)
Specificity – in the clinical context, clinical specificity is a measure of how frequently an assay will correctly identify a sample as negative for a particular disease or state, i.e. how well an assay identifies the analyte to the exclusion of other molecules that are not of interest. It is calculated as:
Specificity = True Negatives / (True Negatives + False Positives)
Cross-reactivity - can be described as a measure of how many non-target molecules are picked up by the assay, and therefore affect the output of the assay, or it can sometimes be used to describe the extent to which an assay identifies multiple target analytes.
Interference - Interference is a broad-brush term that essentially refers to any effect that can have an impact on the assay reading. For example, alcohol in the blood sample of an individual may have an effect on the performance of the assay or certain other molecules may interfere with the analyte, preventing it from being detected in the assay, or causing the assay to give the wrong result.
Precision - Precision is a measure of how often the assay will give the same answer for a given sample i.e. its repeatability. Intra-assay precision describes the variation in the response for a given sample within a single run, and inter-assay precision describes the variation in the response for a given sample across multiple independent runs of the same assay. The precision of the same assay between different laboratories is also an element of inter-assay precision. The higher the variation, the lower the precision and vice versa. Precision is most often represented as percentage coefficient of variation (%CV) (the coefficient of variation is the standard deviation for repeated readings expressed as a percentage of the mean and indicates the extent of variability in relation to the mean of the population (the higher the coefficient of variation, the higher the variability)), and occasionally as a standard deviation.
Signal to noise ratio - there will always be a low level background signal that is picked up when the assay is run even if there is no analyte present in the sample. The signal to noise ratio relates to how distinguishable the analyte signal is from the low level background signal, or 'noise'. If the background noise signal range for a number of blank samples is wide, then a higher positive signal from the sample (which can be achieved by less sample dilution) is required to ensure that any signal being generated by the analyte is distinguishable from the background signal compared to if the noise signal range from blank samples is tight.
Accuracy – Accuracy describes how close the average measured value is to the true value for a sample, as determined by an external source such as a reference method or a certified reference material.
Stability - The stability of the samples that are to be analysed and the reagents/components of the assay is essential to a well-performing assay. Degradation can result in misleading readings.
- As in many cases, it is a good discipline to consider what the cited prior art teaches the Skilled Team in the absence of the Patent, so I consider the teaching here.
- Gorelik is a Biogen paper that was published in the Annals of Neurology in 2010 entitled "Anti-JC Virus Antibodies: Implications for PML Risk Stratification". It reports on a study that was undertaken to establish an ELISA for the detection of anti-JCV antibodies in MS patients and evaluate the potential utility of that assay for identifying patients at higher or lower risk of developing PML.
- Gorelik describes a two-step assay comprising an ELISA (an indirect ELISA) and a supplemental confirmation test (a competitive ELISA):
i) First step: The ELISA produces an nOD readout that is related to the anti-JCV antibody level of the sample. Samples with nOD levels <0.1 are considered anti-JCV antibody negative; samples with nOD levels >0.25 are considered anti-JCV antibody positive, and samples with nOD values between 0.1 and 0.25 are classed as indeterminate and subject to the supplemental confirmation test.
ii) Second step: The supplemental confirmation test produces a percentage inhibition figure. Samples exhibiting >40% inhibition are classified as anti-JCV antibody positive and those with ≤40% inhibition are classified as negative.
- By the priority date, the Skilled Neurologist reading Gorelik would have known about the Biogen STRATIFY JCV assay that had been introduced in 2011 (this is not the same as the current STRATIFY JCV DxSelect assay) and would have considered it likely that Gorelik was describing that assay. However, whereas the results of the STRATIFY JCV assay were reported to clinicians in binary form - a sample was reported as either JCV antibody-positive or JCV antibody-negative - Gorelik discloses that the assay measures the level of anti-JCV antibodies in a patient sample expressed as an nOD value (although Dr Molyneux stressed in his second report that Gorelik uses the assay solely to determine whether a sample is seropositive or seronegative).
- The introduction notes the link between JCV infection and PML in immunosuppressed patients and theorises that serologic detection of anti-JCV antibodies may be a sensitive means to detect current or past infections with JCV with a view to risk stratification.
- Gorelik is divided into three main sections: "Patients and Methods", "Results" and "Discussion". There is also Supplemental Data which includes further technical information relating to the two-step assay.
- The authors tested plasma, serum and urine samples from 831 patients in the STRATA cohort who had received intravenous natalizumab every 4 weeks for 48 weeks. They reported that since the marketing approval of natalizumab in June 2006 down to July 2010, there had been 58 confirmed cases of PML on natalizumab treatment (and 3 PML cases in the pre-approval clinical trials). Pre-PML samples were only available from 17 of the 58 confirmed cases of PML.
- Details of the assay used to detect antibodies to the JCV virus like particles were provided in the Supplemental Data. Sandoz were keen to draw attention to the following 'key features' noted by Mr Scrimshaw:
i) The capture antigens used in the ELISA were JCV MAD-1 virus-like particles ("VLPs"). Gorelik cross-refers to a paper by Sunyaev on the production of VLPs.
ii) Two positive controls and a negative control were prepared by pooling human sera (a CGK method to the Skilled IDS). The reactivity (measured as OD) of the two positive controls was around 1 and 0.25 respectively. The reactivity of the negative control was similar to the assay buffer (although details of the assay buffer are not provided). No further details of the controls are provided.
iii) The results of the ELISA were reported as nOD values calculated by dividing the mean OD of sample replicates by the mean OD of positive control replicates. Gorelik explains that the results were normalised to enable "comparison of results between assay plates, assay runs, and analysts". Gorelik does not specify which positive control was used to produce the normalised OD (i.e. the high or low positive control). Mr Baldwin says that the Skilled IDS would understand that the high positive control was used for normalisation, while Mr Scrimshaw says that the Skilled IDS may think that more likely, but no definitive information is provided.
iv) The supplemental confirmation test was a competitive ELISA used to distinguish patients with JCV-specific antibodies from those with low affinity antibodies and/or antibodies that were cross-reactive to denatured antigen or other common polyomaviruses (such as BKV). The output of the supplemental confirmation test was a % inhibition level; a higher % inhibition level was indicative of JCV-specific antibodies.
- Mr Scrimshaw's evidence was that details about both steps of the two-step assay are missing from Gorelik. For the first step, whilst the Skilled IDS could make a functioning assay that adopted the same general format of the Gorelik assay and measured anti-JCV antibody levels based on nOD values, that assay would generate different nOD values to the Gorelik assay and would have different cut points. His view was that there is even less information in Gorelik about the second step.
- I have to return to these topics when I come to consider aspects of Sandoz's insufficiency case.
- Gorelik used plasma and serum samples from 831 MS patients who were being treated with natalizumab in the STRATA study as well as from 17 patients who had developed PML. The samples from the PML patients were taken at various time points before PML diagnosis.
- Fig. 1 is a plot of anti-JCV antibody levels in MS patients who are either urinary JCV DNA positive (and therefore known to be infected with JCV) or urinary JCV DNA negative (and therefore of uncertain JCV-infection status). As no urinary JCV DNA positive patient showed ELISA reactivity below an nOD of 0.1, Gorelik selects this as the low cut point such that patients with an nOD <0.1 are determined to be JCV negative. This is the point at which the empirical false-negative rate is 0%.
- However, as Gorelik explains, using a cut point that controls the false negative rate at 0% (i.e. nOD 0.1) is unlikely to exclude detection of antibodies that cross-react to denatured antigen or other common polyomaviruses (i.e. false positives). Accordingly, Gorelik discloses use of a supplemental confirmation test to distinguish patients with JCV-specific antibodies from those with low-affinity and/or cross-reactive antibodies. Fig. 3 is a plot of nOD (derived from the ELISA) against % inhibition (derived from the confirmation test) and shows that the vast majority of urinary JCV DNA positive patients have a percentage inhibition above the 40% level. Accordingly, Gorelik designates 40% inhibition as the cut-off between JCV-positive and JCV-negative patients in the supplemental confirmation test. Gorelik defines an nOD of 0.25 as being the higher end of the indeterminate zone on the basis that at nOD values >0.25, the probability of observing >40% inhibition in the supplemental confirmation test was approximately 95%.
- Prof Berger's evidence was that the Skilled Neurologist would note from Fig. 3 that there was a broad spread of nOD values above the positive threshold (nOD value of 0.25) for both urinary JCV DNA positive and urinary JCV DNA negative patients, indicating variability in the levels of anti-JCV antibodies amongst JCV positive patients.
- 204 of the 831 patients tested were "urinary JCV DNA positive", establishing a reference population of patients known to have been infected with JCV. The range of anti-JCV antibody levels in the serum of the urinary positive population was then compared to those of patients in the urinary negative population (who may or may not be infected with JCV). This comparison is illustrated in Figure 1 on page 297.
- The data shows that overall anti-JCV antibody levels in the positive reference population tended to be higher, but the range of anti-JCV antibody levels in the urinary negative population was broader, and that the two populations overlapped.
- Figure 2 illustrates the relationship between viral levels and antibody reactivity in the urinary positive population. The authors conclude (page 297 left hand column):
"These data suggest that seronegative results are likely due to an absence of JCV infection, rather than to very low viral levels".
- Dr Molyneux produced this annotated version of Figure 3B of Gorelik, which gives results for the urinary positive cohort, as follows:
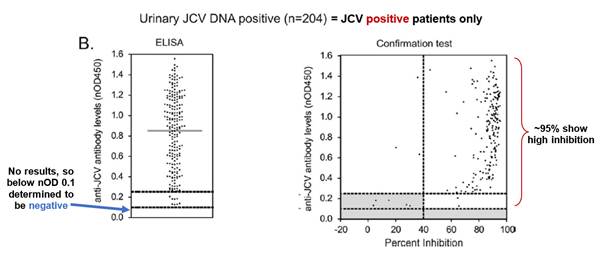
- Consistent with Figure 1, the lefthand graph shows that no urinary positive patients had antibody levels below nOD 0.1. This creates a threshold below which patients are considered to be anti-JCV antibody negative. The righthand graph shows that the vast majority of samples from urinary positive patients showed high inhibition in the confirmation test (and therefore true anti-JCV antibodies).
- The authors draw a conclusion with respect to this 2-step assay on page 298 stating that by combining the ELISA and confirmation test the chance of detecting samples with JCV-specific antibodies is greatly enhanced. They state:
"In the final analysis, samples with nOD450 values < 0.10 in the ELISA are considered anti-JCV antibody negative, and those with nOD450 values > 0.25 in the ELISA are considered anti-JCV antibody positive. Samples with reactivity between nOD values 0.10 and 0.25 are further evaluated in the confirmation test. In the confirmation test, all samples exhibiting >40% inhibition are classified as anti-JCV antibody positive (see Fig 4). At nOD450 values >0.25, the probability of observing >40% inhibition was approximately 95%."
- Having defined the cut-points, the authors use the results of the assay to estimate the seropositivity for anti-JCV antibodies in the STRATA population as 53.6% (subject to confidence limits).
- Gorelik also analyses the result of the 204 urinary JCV DNA positive patients (i.e. those patients known to be infected with JCV) and finds that just 5 fall within the indeterminate range on the ELISA (i.e. have nOD values between 0.1 and 0.25) and demonstrate ≤40% inhibition in the confirmation test. This demonstrates that the false-negative rate of the two-step ELISA is 2.45% (i.e. 5 out of 204).
- Under the next heading, the authors describe the longitudinal study they carried out to assess the stability of anti-JCV antibody status over time and as regards demographics of the cohort. Seroconversion and reversion were studied in a population of 296 patients for whom serum samples were serially collected over a period of five years. 43% were observed to be stable seropositives and 39% stable seronegatives. Approximately 2% per annum converted from anti-JCV antibody negative to positive ("seroconversion") and 3% over five years converted from anti-JCV antibody positive to negative ("seroreversion").
- This study appears to identify differences in seropositivity rates depending on gender, age and geographical location, as shown in Figure 5. The sample sizes are small.
Anti-JCV Antibodies in PML Patients
- Finally, Gorelik considers "whether detection of anti-JCV antibodies using our validated 2-step assay has value for assessing PML risk". Serum samples from 17 patients who subsequently developed PML were tested using the two-step assay. The nOD value, percentage inhibition in the confirmation test and timing of sample collection prior to PML diagnosis for these patients was summarised in Table 1 and shown in fig 6B:
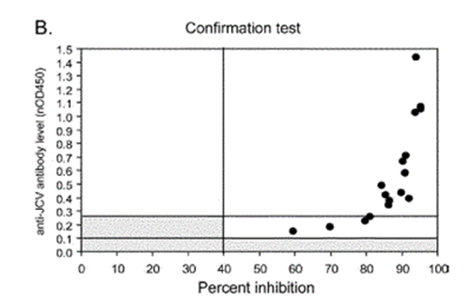
- Gorelik notes that 100% of the samples from patients who developed PML were categorised as anti-JCV antibody positive with the two-step assay (14 of 17 samples from patients who developed PML were above nOD 0.25; 3 samples were between nOD 0.1 and 0.25 but each produced over 40% inhibition in the confirmation test), compared to the 53.6% seroprevalence estimated in the STRATA cohort (which would have indicated 9 out of 17).
- The authors stated that (p.300, Column 2):
"Although these data are based on a small number of pre-PML samples, they suggest that patients without detectable levels of anti-JCV antibodies may have a lower risk of PML development compared to patients who have detectable anti-JCV antibodies. Further clinical studies are underway to confirm this and determine whether the assay [the validated 2-step ELISA] may be a useful tool for stratifying natalizumab-treated MS patients for higher and lower risk of PML"
- Having observed a lack of evidence that testing for JCV DNA in blood is useful for predicting PML development, the paper suggests that:
"...serological assessment of anti-JCV antibody status may offer a more sensitive and practical means of identifying patients who may be infected by JCV."
- The authors observe that, in other papers, JCV seropositivity rates have varied:
"The variation in seropositivity rates among these studies is likely due to marked differences in the sample size, demographics, and, perhaps most importantly, differences in assay methods. Our observation of 54% seropositivity is consistent with rates that have been reported in other recent large studies of healthy subjects."
- They then state:
"The overall risk of developing PML in natalizumab-treated patients is rare, and is likely dependent on the convergence of multiple factors within an individual, including infection by JCV. Although the presence of anti-JCV antibodies alone may not be highly predictive of PML risk, our observation that none of the 17 pre-PML samples were classified as seronegative merits further study. Evaluation of the utility of the assay for risk stratification in the context of ongoing clinical studies will help elucidate if the risk may be significantly lower in a seronegative group compared with that of a seropositive group."
- The authors then discuss the low false negative rate of 2.5% and state:
"The false-negative rate in the assay, combined with the ~2% annual seroconversion rate observed in the STRATA cohort, suggests that periodic testing of seronegative patients is warranted in clinical practice."
- The discussion concludes:
"In summary, detection of anti-JCV antibodies using this novel 2-step assay may provide a tool for stratification of patients into groups at higher risk for PML (ie, seropositive) and those at lower risk (ie, seronegative). Because the risk of PML will still be quite low in seropositive patients, it is likely that additional variables, such as the presence of viral mutations, host factors (eg, genetics and immune status), and previous therapeutic interventions may contribute to development of PML. Approaches to further stratify the risk of PML in the anti- JCV antibody positive group are being actively explored in ongoing clinical studies in the United States and in Europe. In addition, we continue to optimize the current anti-JCV antibody assay to help reduce the false-negative rate. For the time being, clinical vigilance remains the primary tool for early detection of PML. However, the clinical management of MS patients using or contemplating the use of natalizumab could be facilitated by tools, such as this assay, that may ultimately allow for a better understanding of a patient's individual risk of this serious adverse event."
- Prof Berger noted that the data presented in Table 1 and Fig. 6 shows that generally the PML patient samples had nOD values in excess of 0.25, many being considerably in excess of 0.25, and that all had percentage inhibition values of about 60% or higher.
- Prof Berger's view was that the Skilled Neurologist would regard the data as giving the impression that PML could be associated with higher nOD values and higher percentage inhibition values. As Sandoz said, this was an impression only: as Prof Berger pointed out the sample size (17 PML patients) was too low to draw a conclusion and as the samples tested were collected at varying time points in advance of PML diagnosis they did not necessarily reflect the position at the time of or in the run up to diagnosis. Nevertheless, Prof Berger considered that the idea of stratifying risk according to antibody level would have been obvious to the Skilled Neurologist reading Gorelik.
- Dr Molyneux disagreed. He said the Skilled Neurologist would not gain this impression from the data. He said the only conclusion which could be drawn was that all 17 samples were anti-JCV antibody positive, an entirely unsurprising point given that prior exposure to JCV was a known risk factor associated with developing PML. He said there was otherwise no clear pattern in the data. He also pointed out that the data in Fig 6 and Table 1 relate only to PML samples which, without a direct comparison against the non-PML patient population tested, would not allow the Skilled Neurologist to draw conclusions regarding an association between nOD and PML.
- Biogen contended this impression formed by Prof Berger could only have come about through hindsight. They pointed out he learned of the invention over a decade ago and submitted it is clearly ingrained into his thinking about PML.
- In cross-examination, I agree that Prof Berger was unable to provide a meaningful explanation of how he formed the impression from Figure 6 that anti-JCV antibody levels seemed higher in PML patients than non-PML patients. He appeared surprised to learn that the median antibody level of that population (nOD 0.435 - entry 10 in Table 1) was lower than even a generous estimate of the median in the antibody positive population discernible from Figures 3A and 3B (put in cross-examination as nOD 0.5, coming down from the approximately 0.8 for the urinary positive population shown in Figure 3B). In those circumstances, Biogen submitted that the only sensible explanation for his view is hindsight. Indeed, they said he appeared to come very close to accepting this at p.637, lines 8 - 12:
8 A. Again, it is probably not the table alone, but also the fact
9 if you are measuring antibodies in a group which you then
10 label as potential risk group, then I think it is intuitively
11 to get the impression the higher the antibodies, the higher
12 the risk.
- On his own evidence, his 'intuition' was not part of the CGK. For that reason Biogen submitted it is very hard to see how Professor Berger can have sought to ascribe such intuition to the Skilled Neurologist without hindsight.
- In their closing, Sandoz clearly backed away from the 'impression' argument. The argument was not dropped but it was clear that Sandoz placed greater emphasis on their back-up argument which I consider below. For present purposes, I find the Skilled Neurologist would not form Prof Berger's impression from Gorelik. I must also keep in mind Biogen's point that he could only have formed this impression using hindsight.
- WO369 is a Biogen patent application published in July 2011 entitled "Assay for JC Virus Antibodies". Gorelik was one of the inventors. The Abstract states that: "the disclosure relates to methods and reagents for analyzing samples for the presence of JC virus antibodies...In one embodiment, determining the level of anti-JCV antibodies in the subject sample provides a method of identifying PML risk in a subject".
- Although I must assess each piece of prior art on its own merits, it was common ground between the experts that much of the disclosure of WO369 is similar to that of Gorelik, e.g. Figs. 1, 2, 4 and 5 of WO369 are the same as Figs. 1, 2, 3 and 4 of Gorelik, while Fig. 6 of WO369 is similar to Fig. 6 of Gorelik save that fewer data points are plotted (11 PML patients as opposed to 17 in Gorelik).
- However, as Sandoz submitted, WO369 contains additional information about use of the assay to assess PML risk in patients as well as some additional technical information about the assay. I will largely concentrate on the additional information.
- The invention is summarised on page 2 essentially as follows:
i) A biological sample is obtained from a subject. The sample may be serum, plasma, blood, urine or cerebrospinal fluid among others. The individual sampled may have received immunosuppressant or immunomodulatory treatment or may be suspected of having PML.
ii) The sample is contacted with JCV HPVLPs, which bind anti-JCV antibodies.
iii) The level of antibody binding is measured. WO369 describes that detecting anti-JCV antibodies can indicate a risk of developing PML or other adverse symptoms, which can inform treatment decisions and the type and extent of any monitoring for adverse symptoms.
iv) The level of antibodies bound is correlated with a "reference", such as a control sample.
v) A confirmatory test can be performed as a second step in the assay, measured in terms of inhibition compared to the first step.
- This is the 2-step assay discussed in Gorelik. However, on top of disclosing use of the two-step assay to determine whether a patient is anti-JCV antibody positive or anti-JCV antibody negative, WO369 expressly discloses the following (emphasis added):
i) p.6 ll.4-14 discloses an aspect of the invention that "features a method of identifying PML risk in a subject by determining the level of anti-JCV antibodies in a sample from the subject, such as from a plasma, blood or serum sample; and assigning a risk level to the subject according to the level of anti-JCV antibodies in the sample";
ii) p.6 ll.15-20 discloses an embodiment in which the method of identifying PML risk in a subject includes determining the level of anti-JCV antibodies in the sample of a subject taken at two different time points and comparing them to determine whether the subject is at increased risk of PML at the later date;
iii) p.6 ll.21-27 discloses:
"In one aspect, the invention features a method of monitoring PML risk in a subject, the method comprising determining the level of anti-JCV antibodies in a subject using a sample from a first date; assigning a risk of PML (e.g., high, or moderate or low risk) based on the level of anti-JCV antibodies in the subject on the first date; determining the level of anti-JCV antibodies in the subject using a sample from a second date; and assigning a risk of PML (e.g., high, or moderate or low risk) based on the level of anti-JCV antibodies in the subject on the second date."
- Sandoz were keen to point out some additional technical information provided in WO369 about the two-step assay, in support of their insufficiency allegations. For the moment I simply note their contentions which were as follows:
i) Example 1 sets out a method for the synthesis and purification of Highly Purified VP1 Particles (for use as the antigen in the assay), and Example 7 sets out an alternative method for their purification. As Mr Scrimshaw noted in his first report, WO369 does not explain how the purification method chosen affects the assay and it is not clear from WO369 whether one purification method is preferred over the other.
ii) Example 2 identifies that the positive and negative controls used in the assay have the following target OD and % inhibition values:
Positive Control 1 - target OD of about 1.0, >80% inhibition;
Positive Control 2 - target OD of about 0.25, >80% inhibition;
Negative Control - target OD of approximately 0.07 (assay buffer has an OD of approximately 0.045).
As described in Gorelik, the controls are pooled donor sera.
iii) Example 3 sets out the equation for normalisation of OD and states that it is the high positive control (Positive Control 1) that is used to normalise the sample OD measurements.
iv) Mr Scrimshaw explained what the Skilled IDS would understand WO369 to be disclosing in detail in his first report. As with Gorelik, Sandoz contended that important information is missing in WO369 that means that the Skilled IDS would not be able to reproduce the assay so as to obtain the same cut-off thresholds for positive, negative and indeterminate samples in terms of nOD values or the underlying anti-JCV antibody levels as in the WO369 assay.
- In his first report, Prof. Berger described WO369 as disclosing that patients could be stratified as being at low, moderate or high risk of PML based on their level of antibodies, and that patients in the high and moderate risk categories would be positive for anti-JCV antibodies.
- A suggestion was put to Prof. Berger about p.6 ll.21-27 during cross-examination, namely that "it could be that the high antibody levels are providing protection, whereas the moderate antibody levels are an indication of immunosuppression". Sandoz noted that this suggestion had never been made by Dr Molyneux. The passage does not refer to high, moderate or low levels of antibodies but to high, moderate or low risk of PML based on the level of anti-JCV antibodies in the patient. In any event, the suggestion appears to make no difference. The teaching is linking levels of antibodies to risk of PML. As Sandoz submitted, it does not matter whether it could be read as suggesting that there is an inverse relationship between levels of antibodies and risk of PML. The Skilled Team would still proceed to carry out the same study and look at the nOD results (see further below).
- Prof. Berger's evidence was that "this idea of stratifying risk based on levels of anti-JCV antibodies would have been of substantial interest to the Skilled Neurologist" and that they would want to follow it up "by collecting and reviewing data regarding antibody levels in patients to see whether the statement that patients can be grouped into high, moderate or low risk of developing PML based on their antibody levels was supported by the evidence." Sandoz say he was never actually challenged on that evidence - after cross-examination based on a misreading of the passage, the point put was that there was no support or reasoning given for the statement.
- Dr Molyneux was very dismissive of this disclosure in his written evidence, a view which hardened in his oral evidence:
i) First, in his second report, Dr Molyneux said it was 'not clear' whether the reference to 'level of anti-JCV antibodies' at line 5 was 'a reference to anything other than the method of arriving at the binary positive/negative result using the one-step or two-step assay described elsewhere in WO369'. Although he did not say so expressly, it is reasonable to assume he meant the same would apply to the same expression wherever used on page 6.
ii) Second, as for the passage at lines 21-27, although he quoted this in his first report, noted that it was a difference over Gorelik, and recognised it referred to assigning risk of PML at a first date and includes similar wording for assigning risk on a second date, his only comment was:
'There is no data or reasoning to support a suggestion that risk may be assigned based on any particular level of antibody titre, or what would represent a "moderate" risk of developing PML. Given the absence of reasoning and the brevity of the statement, I do not believe the Skilled Team would attach any weight to it.'
- Thus, Dr Molyneux's written evidence focussed on the absence of data and reasoning and the brevity of the statement, which he said meant that the Skilled Neurologist would not attach any weight to it. In cross-examination he constantly returned to the theme that it was a single word (apparently a reference to the word 'moderate') embedded in a 35-page document which also contained other aspects.
- However, also in cross-examination, Dr Molyneux appeared to agree with Prof Berger [T2 169/13 - 170/5]:
Q. What they are saying here is that you assign a risk of PML which can be high, moderate or low risk, depending on the level of antibodies in the subject, yes?
A. Yes.
Q. That is clearly saying more than just positive negative, is it not?
A. I would agree, it is.
Q. Because if it is high or moderate, it is a stratification of the antibody positive population, at the very least?
A. Well, it is beyond the binary result, that is not in dispute. I am not sure whether high or moderate are both positive, but it is beyond dispute that high, moderate or low is talking about more than just a binary readout.
Q. Yes. We know that the low risk will be people who are negative, presumably, and they are dividing the positives into high and moderate risk?
A. Yes, that is one reasonable assumption of what they have said.
- By the priority date the Skilled Neurologist already knew about STRATIFY JCV and its use to stratify patients into higher or lower risk based on whether they are anti-JCV antibody positive or negative. What was new to the Skilled Neurologist was the idea at p.6 ll.21-27.
- In my view, Dr Molyneux was far too dismissive of the significance of the disclosure of WO369, and his views did not accord with those of the Skilled Team reading WO369 with interest. The Skilled Team would note that WO369 came from the same group at Biogen as Gorelik and would therefore be interested in any disclosure which told them something new over and above Gorelik and/or their existing knowledge about the STRATIFY JCV assay. The disputed passage provided new information, clearly going beyond the simple binary report from the STRATIFY JCV assay - positive or negative. It put into the minds of the Skilled Team the idea of further stratifying the risk of PML based on the level of anti-JCV antibodies in the sample. I agree with Prof Berger that it would be of real interest to the Skilled Neurologist.
- This case involves an unusual issue of interpretation of claim 1. For that reason, it is necessary to summarise a lot of the disclosure in the specification. As Sandoz submitted, the specification is not an easy read since there is much repetition between the 98 paragraphs under the heading 'Summary of Invention' and the paragraphs in the Detailed Description. What I will attempt to do is to identify what appears to matter.
- [0001] explains that the invention relates to methods of assessing a patient's risk of developing Progressive multifocal leukoencephalopathy (PML).
- Under the heading 'BACKGROUND OF INVENTION':
'[0002] The anti-VLA-4 (Very Late Antigen 4) antibody therapeutic natalizumab is indicated to treat relapsing forms of multiple sclerosis (MS) and moderate-to-severe Crohn's Disease. Natalizumab treatment, however, is associated with an increased risk of progressive multifocal leukoencephalopathy (PML), an opportunistic brain infection caused by the JC virus (JCV). PML occurs primarily in immunocompromised individuals and in patients receiving certain immunomodulatory therapies, including natalizumab. PML is hypothesized to be the result of a complex interaction between host and viral factors, leading to reactivation and mutation of latent archetype JCV to a neurotrophic form which can infect oligodendrocytes in the central nervous system.'
- Certain cited prior art are acknowledged in [0003], including both Gorelik and WO369. I mention certain of the acknowledgements which may achieve significance later:
i) Sandrock (an article in 2011 in Neurology) 'relates to the risk stratification for PML in MS patients and the role of prior immunosuppressant use, natalizumab treatment duration and anti-JCV antibody status'.
ii) Gorelik 'relates to anti-JCV antibodies and implications for PML risk stratification'.
iii) WO369 'relates to methods and reagents for analyzing samples for the presence of JC virus antibodies, such as a method that includes obtaining a biological sample from a subject, contacting the sample with highly purified viral-like particles (HPVLPs) under conditions suitable for binding of a JCV antibody in the sample to an HPVLP, and detecting the level of JCV antibody binding in the sample to HPVLP.'
- Then under the heading 'Summary of the Invention' [0004] explains that the invention relates to an optimized analytically validated sensitive assay for detecting the presence of JCV antibodies in serum or plasma. [0005] is a consistory clause for claim 1, which, it will be noted, does not concern itself with % inhibition. In fact, all references to % inhibition are removed from the claims by the amendments.
- [0006] and [0007] introduce 2 additional steps to the method of claim 1, both concerned with % inhibition. These are labelled (c) and (d). Then [0008]-[0010] add the option of providing the results to another entity e.g. a healthcare provider, for various combinations of steps in the methods - (a) & (b), (a), (b) & (c) and (a)-(d). It is apparent that steps (i) and (ii) in [0005] and claim 1 represent steps (a) & (b) in these paragraphs.
- It is not necessary to refer to much of the remainder of this section of the Patent. I pick out certain passages to which reference was made during the trial. First, [0033]-[0036]:
'[0033] The methods disclosed herein are based at least in part on the discovery that anti-JCV antibody titer and other characteristics such as affinity/avidity can be indicators of a patient's risk of developing Progressive Multifocal Leukoencephalopathy (PML).
[0034] Accordingly, the invention features, a method of evaluating a patient's risk of developing PML, comprising acquiring knowledge of a JC Virus (JCV) antibody titer (e.g., determined as described herein and expressed as normalized optical density (nOD) or index) as defined in the claims.
[0035] In one embodiment, an anti-JCV antibody titer or percent inhibition is determined in a biological sample from a patient, such as a blood (serum or plasma), or CSF sample.
[0036] If the titer or/and percent inhibition, or a function of both values is determined to be below a pre-determined level, the patient is determined to be at a lower risk of developing PML, and if the titer and/or percent inhibition, or a function of both values is determined to be at or above the pre-determined level the patient is determined to be at a higher risk of developing PML.'
- In cross-examination, Mr Scrimshaw accepted that the Patent is explaining that it is antibody titer, expressed as an index value, that is important in evaluating a patient's risk of PML, and that the level of risk of PML depends on whether the titer is above or below a predetermined level (see [T2 177/13- 178/16])
- Next, [0044]-[0045] and [0060]:
'[0044] The patient can be determined to have a lower risk of PML if the anti-JCV antibody titer as indicated by index value or nOD is determined to be < 0.5, the patient can be determined to have a higher risk if the anti-JCV antibody titer as indicated by index value or nOD is determined to be > 0.5 and <1.5. The patient is determined to have an even higher risk if the anti-JCV antibody titer as indicated by index value or nOD is determined to be > 1.5.
[0045] If the assay indicates that the biological sample does not contain JCV antibodies above a background level, the patient can be determined to be at lower risk for PML.'
'[0060] The patient is determined to have a higher risk of PML if, (i) the anti-JCV antibody titer as indicated by index value or nOD is determined to be > 1.5 and the percent inhibition value is determined to be > 70%,'
- [0073]-[0075] are relevant to the issue over claim 8:
'[0073] A patient who has received an anti-VLA-4 therapy, such as natalizumab, for longer than 24 months (e.g., for 25 to 48 months, such as 26, 30, 36, 42 or 48 months or longer), and who is determined to be positive for JCV, can be determined to be at a higher risk for PML. The patient can accordingly be determined not to be a candidate to receive further treatment with an anti-VLA-4 therapy, or can be determined to be a candidate to receive treatment with an anti-VLA-4 therapy accompanied by more frequent monitoring.
[0074] In an embodiment, a patient who has received an anti-VLA-4 therapy, such as natalizumab, for longer than 24 months (e.g., for 25 to 48 months, such as 26, 30, 36, 42 or 48 months or longer), and who has not received prior treatment with an immunosuppressant (other than an anti-VLA-4 therapy), and who is determined to be positive for JCV, and is determined to be at a higher risk for PML. For example, the patient can be determined to have a risk of PML of 4/1000 patients. The patient can accordingly be determined not to be a candidate to receive further treatment with an anti-VLA-4 therapy, or can be determined to be a candidate to receive treatment with an anti-VLA-4 therapy accompanied by more frequent monitoring.
[0075] A patient, e.g., an MS patient, who has received prior treatment with an immunosuppressant other than an anti-VLA-4 therapy, and who is determined to be positive for anti-JCV antibodies, or JCV nucleic acid, can be determined to be at a higher risk for PML. ...'
- The Detailed Description begins with a (long) introductory section from [0104]-[0118]. I pick out those passages which are pertinent. The section begins with these statements:
'[0104] The invention is based, at least in part, on the discovery of new and improved methods of assessing the risk of a patient for PML that include assessing anti-JCV antibody titers or percent inhibition. The invention is based at least in part on the discovery that anti-JCV antibody titer and percent antibody inhibition can be an indicator of a patient's risk of developing Progressive Multifocal Leukoencephalopathy (PML).
[0105] Applicants have also developed an optimized assay for determining anti-JCV antibody titer levels in a biological sample, and a method for assaying the antibodies qualitatively by determining percent inhibition values, and using this information to determine the risk of a patient for developing PML. ...'
- Paragraph [0111] again makes reference to three known risk factors for PML (duration of natalizumab treatment, prior immunosuppression and JCV serostatus) independently contributing to PML risk:
'The ability to identify subpopulations of patients at distinctly different PML risks allows for better characterization of risk than previous methods (i.e., overall PML risk) and should assist healthcare professionals and patients in making more informed benefit-risk treatment decisions.'
'[0116] JCV antibodies are detected by an ELISA assay. In one embodiment, JCV antibodies can be detected by the method described in International Application Number PCT/US2011/20832, which utilizes HPVLPs under conditions suitable for binding of an anti-JCV antibody for detecting the level of anti-JCV antibody binding in a biological sample. Methods of determining JCV status also include methods of determining anti-JCV antibody titer and percent inhibition. Detection of anti-JCV antibody titer and percent inhibition typically include a two-step antibody detection assay as described in International Application Number PCT/US2011/20832.' [This is WO369].
- A series of definitions and explanations of terms are set out in [0119]-[0188]. In that section, the HPVLPs are discussed from paragraph [0140] to [0151]. It is explained at [0140] that in general the VP1 capsid protein from the MAD-1 strain of JCV is used.
- Paragraph [0187] states that:
"PML risk stratification tools are useful as one component in making individual benefit-risk treatment decisions for patients taking or considering taking a VLA4 inhibitor or other therapeutics known to increase risk of developing PML. Quantification of a patient's PML risk can be used, for example, in benefit-risk analysis."
- The specification then turns to Examples 1-7 which in the usual way are said to further illustrate but not further limit the invention.
- Example 1 is a quantification of PML risk in MS patients based on prior immunosuppressant use, duration of treatment and anti-JCV antibody status (i.e. positive or negative). Anti-JCV antibody status was determined using the two-step ELISA disclosed in Gorelik also referred to in the Patent as the "Gen1 assay". The study is based on data from 5,896 patients, from three different clinical studies conducted by Biogen (AFFIRM, STRATIFY-1 and TYGRIS-US) and the Swedish national registry. Figures 1A and 1B illustrate the effect of natalizumab treatment duration on estimated PML incidence per 1000 patients. These figures indicate that PML risk generally increased with duration of natalizumab exposure. Table 1 compares prior immunosuppressant use in natalizumab-associated PML patients, compared with MS patients on the TYGRIS-US study. PML patients were more likely to have received immunosuppressant therapies, and for a longer average duration. Figure 2 then illustrates the combined effect of these two risk factors on PML incidence, separating the patients tested into four subgroups for the presence or absence of each risk factor and illustrating their estimated incidence of PML.
- Fig. 3 is a flow-chart summarising the PML risk status (in terms of incidence per 1000 patients) associated with the various risk factors. Prof. Berger's evidence is that the results of Example 1 would already have been CGK to the Skilled Neurologist, having been reported in the Kappos review paper. Sandoz submitted that therefore Example 1 does not disclose anything new to the Skilled Team, either in terms of the assay or methods for stratifying risk.
- Sandoz nonetheless drew attention to the point that the risk of PML for an anti-JCV antibody positive patient, with no prior immunosuppressant use whose duration of treatment with natalizumab is less than 2 years, is 0.35 / 1000 patients (Fig. 3). Such a patient has only one of the three known risk factors for PML yet, according to [0079] of the Patent, is an example of a patient at "higher risk" of PML.
- Paragraph [0199] explains that samples from 5,896 MS patients were available for testing for anti-JCV antibodies (summarised in Table 2), and that the overall seroprevalence in the MS population assessed was 55%. Paragraph [0201] describes that 100% of the 25 samples tested from patients who developed PML (summarised in Supplemental Table 1) were positive for anti-JCV antibodies.
- Paragraphs [0203] to [0206] and Tables 4-5 describe the analysis carried out to calculate the estimated incidence of PML according to JCV serostatus and duration of natalizumab treatment, to demonstrate the increased risk in the antibody positive population. Paragraphs [0207] to [0209] then describe a "combined, quantitative PML risk algorithm" for the estimated incidence of PML according to the presence or absence of the three risk factors, the results of which are illustrated in Figure 3. The authors also describe a "sensitivity analysis" for the estimates in Figure 3, illustrated in Figures 4A and 4B.
- Example 2 is a brief description of how "a novel 2-step enzyme-linked immunosorbent assay (ELISA)" was validated in clinical laboratories. Reference is made to WO369 and the Skilled Team would be likely to understand this assay to be that commercialised as the STRATIFY JCV assay - also referred to as the Gen1 assay in the Patent.
- Example 3 is headed 'A refined two-step JCV assay (the Gen2 Assay) provides more accurate results than the original assay (Gen1 Assay)'. It reports a study of the clinical agreement between the Gen1 assay and what is described as a "new assay" - the "Gen2 assay". [0214] discloses that the Gen2 assay is a modification of the Gen1 assay "following optimization rounds" and goes on to disclose that:
"The [Gen2] assay differs from the [Gen1] assay in at least the follow [sic] ways..."
- There follows a list of six bullet points of difference between the Gen1 and Gen2 assays. Sandoz were very keen to highlight the fact that this list is presented as non-exhaustive. I consider it is necessary to have regard to the first five differences identified as well as the sixth. It is convenient to number the bullet points as follows:
i) 'HPVLP is used at a substrate concentration of 0.4 μg/mL on plates in the first step, and in solution in the confirmatory assay, as opposed to 1 μg/mL used in the Gen1 assay.'
ii) 'Patient serum is diluted 1:101 prior to applying to HPVLP on plates in the first step of the assay, or to HPVLP in solution in the confirmatory assay, as opposed to 1:200 in the Gen1 assay.'
iii) 'The secondary reagent (anti-human IgG) conjugated to HRP is typically diluted 1:20,000 (but may have to be readjusted for new lots to match signal to previous lot), and the incubation time with the conjugate is only 30 min. In the Gen1 assay, the same reagent was diluted 1:80,000 and incubation time was 60 min.'
iv) 'The binding reaction is assayed by incubating the HRP substrate TMB for 20 minutes ± 2 minutes, whereas in Gen1, the TMB incubation was for 20 minutes ± 5 minutes.'
v) 'In the confirmation assay, 10 μL of sample is added to 1 mL of confirmation buffer (1:101 dilution), and the reaction proceeds for 10 to 20 minutes. In the Gen1 assay, a 2x concentration of sample (1:100 dilution) and HPVLP (2 μg/mL) was mixed in equal proportion and then incubated for 60 minutes.'
vi) 'The cut-off calibrator (CO) is adjusted to have a reactivity index of about nOD 1.0, and a positive control (PC) is adjusted to have a reactivity index of about nOD 1.3). The CO and PC are made by mixing an anti-JCV antibody positive serum and an anti-JCV antibody negative serum. For the negative control (NC), which is typically bottle negative sera, the reactivity index target is about 0.1;. Qualitatively, the controls come from different pools of human serum, but from an assay target concentration, they are similar to the Gen 1 control levels.'
- Mr Baldwin treated this list as identifying all the differences between the assays (so for example he says that the HPVLPs used in the Gen2 assay are the same as those used in the Gen1 assay), but in fact the list is plainly non-exhaustive. Quite what is meant by the final sentence of the last bullet point was in dispute and I address that later.
- [0215] & Table 6 set out the results of the study comparing the clinical results obtained in the Gen1 and Gen2 assays at three different sites. The assays show fairly good agreement (94.9% - 100%) in classifying samples as positive for antibodies to JCV but lower levels of agreement (85.7% - 91.8%) in classifying samples as negative for antibodies to JCV.
- At this point, it is necessary to clear up a mini-dispute over what the Patent meant when it uses the word 'titer'. Mr Scrimshaw explained that "titer" can mean the dilution factor that is required before an ELISA signal drops below a predefined threshold, but also can be used more loosely to refer to the level of antibodies in a sample. It was common ground that (save in Figures 5-10 and [0220]) the Patent was using "titer" in that latter sense. As Mr Baldwin said:
"In the Patent, "titer" is not used in the technical sense that Mr Scrimshaw refers to, but rather in the looser sense he mentions of referring to the level of antibodies in a sample".
- Hence, Mr Baldwin said that the Patent involved generating an index value which was "indicative of the level of [anti-JCV] antibodies in the patient sample" and that claim 1 involved producing "an index value as an expression of anti-JCV antibody levels in a sample". At one point in his cross-examination, Mr Baldwin started to suggest that "titer" was being used interchangeably with "nOD" in the Patent. However, he later accepted that what he had said in his reports was correct, and that the Patent was using "titer" to refer to levels of antibodies. By contrast, the index value or nOD is a way of expressing the underlying level or titer of antibodies, as Mr Baldwin had said in his reports.
- Likewise, Dr Molyneux said that the index value was "a numerical indication of a patient's anti-JCV antibody level". He accepted that the Patent was about identifying a patient's anti-JCV antibody level, represented by an index value, and identifying a patient as being at high risk of PML if their anti-JCV antibody level was above a certain threshold, being that represented by an index value of 1.5.
- Notwithstanding the fact that both Mr Baldwin and Dr Molyneux had described the index value or nOD as an indication of the level of antibodies in the sample, there were repeated and rather bizarre attempts during the cross-examination of Mr Scrimshaw to suggest that the Patent was not trying to "communicate a specific level of antibodies". It was common ground that the Patent does not disclose what the level of antibodies is that marks the clinical cut-off. Sandoz contended that the Patent does not disclose information which enables the Skilled Team to identify that or produce an assay with that clinical cut-off (which is the basis of two of their insufficiency arguments). But plainly the Patent teaches that there is a level of antibodies which provides the clinical cut-off.
- I have already referred to the important paragraphs [0033]-[0036] at [186] above, and Mr Scrimshaw's acceptance that it is antibody titer, expressed as an index value, which is important in evaluating a patient's risk of PML.
- The examples which are of significance to this issue are Examples 4, 6 and 7. Sandoz pointed out that in his written evidence Mr Baldwin ignored Examples 4 and 7 and focussed entirely on Example 6. So I will pick up my discussion of the Patent with Example 4.
- Example 4 reports on a study into whether anti-JCV antibody status can be used to categorise a patient's risk for PML. At [0216] the Patent explains that:
"We hypothesized that anti-JCV antibody positive patients could be further stratified for the risk of developing PML based on anti-JCV antibody titers (nOD or index) and anti-JCV antibody avidity/affinity (% inhibition). This hypothesis was derived from the observation that patients having an anti-JCV antibody titer and % inhibition below a predetermined level ("a clinical cut-point") are at lower risk for developing PML compared to the overall anti-JCV antibody positive population."
- The hypothesis is that risk of developing PML can be stratified according to anti-JCV antibody titer, expressed as nOD or index value (and also according to % inhibition).
- [0217] states that to test the hypothesis pre-existing data from both Gen1 and Gen2 assays was collected and analysed, which data included anti-JCV antibody titer information expressed as nOD or index.
- The rest of Example 4 focusses on the findings based on the Gen1 data. Example 6 (dealt with below) addresses the findings based on the Gen2 data.
- [0218] discloses that in the study using the Gen1 assay:
i) There were 356 anti-JCV antibody positive patients and 38 PML patients.
ii) 22% (77/356) of anti-JCV antibody positive patients had nODs >1.0.
iii) 34% (13/38) of patients who went on to develop PML had nODs >1.0.
- The patentee concludes from this data that ~ 1.5 fold more PML patients had an nOD >1.0 compared with non-PML patients, translating to a 2-3 fold risk ratio associated with an nOD >1.0. In other words, this proposes a stratification of risk according to anti-JCV antibody titer, based on the fact that the ratio of PML to non-PML patients is higher above an nOD of 1.0 than below it. As Sandoz pointed out, here it is the titer which matters, not the proportion of the seropositive population that is above the cut-point (and that proportion is said to be 22%, nothing like 50%).
- [0219] discloses the results of a comparison of data taken from longitudinal samples collected from both PML patients (where samples were taken before PML diagnosis) and non-PML patients. 6% of non-PML patients demonstrated a >2 fold increase in nOD over time but the majority of PML patients demonstrated such an increase. The patentee concludes from this that "patients who do not exhibit a significant change in JCV titer over time are at a lower risk of developing PML".
- Fig. 11 is a plot of nOD against % inhibition for non-PML and PML patient samples. As Prof. Berger noted, Fig. 11 does not appear to correspond completely to [0218] as there are only 28 PML patient data points with 8 (29%) having an nOD above the 1.0 threshold. Sandoz provided a version of Fig. 11 with the PML samples coloured in red:
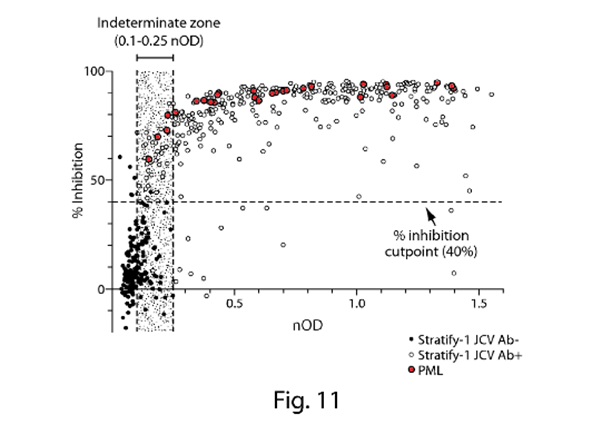
- Reference Example 5 is a study assessing anti-JCV antibody titer changes prior to and after initiation of treatment with natalizumab. Samples from 861 MS patients were analysed, including 5 PML patients. The assay described in Gorelik was used to determine nOD levels. The Patent discloses that whilst only 5% of the anti-JCV antibody positive patients demonstrated a change in nOD values above 0.151, all 5 of the patients who went on to develop PML demonstrated such a change in nOD.
- After treatment was initiated the anti-JCV antibody levels reportedly remained "relatively stable" with a mild decline in nOD in the population of MS patients without PML whereas, in the PML patients, levels of anti-JCV antibodies (nOD) in serum increased at the time of PML diagnosis compared with the baseline values. These increases are detailed in Table 9.
- Example 6 is a study to determine a clinical cut-off, distinct from an analytical cut-off, for delineating high and low risk groups among anti-JCV antibody positive patients such that "a patient's risk of PML would be initially based on baseline anti-JCV antibody titer levels".
- Using the Gen2 assay, samples from 1044 non-PML patients and 38 PML patients (prior to diagnosis), were tested to determine index value and percent inhibition. Antibody titres in the PML patients were measured more than 6 months prior to diagnosis. The fact that samples from 38 PML patients were used in both this study and the study in Example 4 suggests that the patients are likely to have been the same. The results were plotted in Fig. 12, shown below with the PML samples coloured in red:
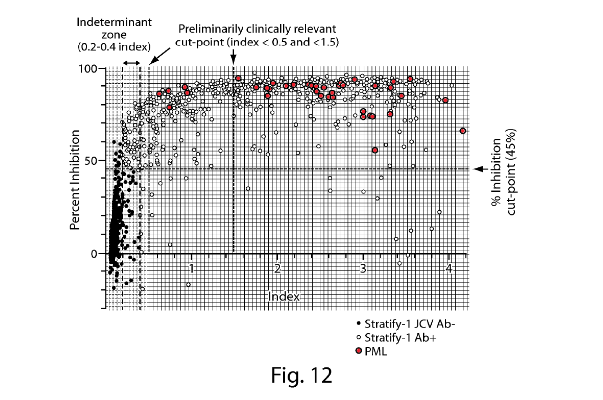
- As Mr Scrimshaw observed in his first report, the results from the Gen2 assay shown in Fig. 12 are very different from those from the Gen1 assay shown in Fig. 11. As a result, Sandoz submitted that while differences would be expected because of the different populations studied, that would not be expected to produce such large differences, and in any event the PML patient samples may well have been the same, so there must be differences in results because of differences in the assays.
- Fig. 12 shows cut-points for % inhibition (at 45%) and for index values at 0.2 and 0.4. In addition, two "preliminarily clinically relevant cut-points" are indicated at index values of <0.5 and <1.5. The reasoning for those cut-points is set out at [0230]-[0231].
- Since various of the arguments on interpretation derive from particular aspects of Example 6, I will set out the four paragraphs in the Patent which describe it (with emphasis added):
[0229] Results from the Stratify I study were used to determine a clinical cut-off distinct from an analytical cut-off to delineate high and low risk groups among anti-JCV antibody positive patients. Thus, a patient's risk of PML would be initially based on baseline anti-JCV antibody titer levels. The Generation II anti-JCV antibody assay was used in this study.
[0230] TYSABRI® non-PML patients (Stratify I, n= 1044) and PML patients (> 6 months prior to PML diagnosis (n=38) were evaluated (FIG. 12). In the Generation II assay, 17% of anti-JCV antibody positive patients had titers (index) below the lowest titer (index) observed for samples from PML patients collected >6 months prior to PML diagnosis, suggesting that those patients may have lower risk for developing PML (like anti-JCV antibody negative patients). Additionally 50% of anti-JCV antibody positive patients had titers (index) below index 1.5, compared to only 13% PML patients from whom samples were collected >6 months prior to PML diagnosis had index <1.5. Also, only 4.4% of the PML patients not known to previously receive immunosuppressant agents had samples with index <1.5, suggesting that those patients may have lower risk for developing PML compared to patient5s [sic] with high anti-JCV antibody titer (nOD or index).
[0231] Patients having an nOD < 0.5 (109/1044 (10.4% of total samples) or 109/549 (20% of anti-JCV antibody positive patients)) were determined to be in the lowest PML risk group (potentially as low as anti-JCV antibody negative patients), as no PML patients had an index<0.5. Patients having index >0.5 but <1.5 were determined to be in the lower risk zone, as 50% [it was common ground that this is an error and should read 30%] of non-PML anti-JCV antibody positive patients and only 13% of PML patients, respectively, had samples in this zone. Additionally, only 4% of PML patients who were not known to receive prior immunosuppressive therapies, had samples with index <1.5 (FIG. 13). Patients having an index >1.5 (271/549 (50%) of anti-JCV antibody positive population) were determined to be at higher risk for PML. Forty seven percent of patients were anti-JCV antibody negative.
[0232] Post-PML diagnosis, patients are subjected to immune-adsorption (IA) or plasma exchange (PLEX) to remove circulating natalizumab and to restore immune function. The anti-JCV antibody levels are rapidly restored to pre-procedure levels in these patients.
- Fig 13 is a scatter plot showing the spread of index values (nOD) observed for the various groups indicated in each column: all non-PML patients, all antibody positive non-PML patients, all PML patients, PML patients who had not received previous immunosuppressant treatment and PML patients who had received previous immunosuppressant treatment. As I understand it, the horizontal lines in the PML columns indicate the median index value for the group concerned, whereas, as the rubric indicates, the index value of 1.5 represents the median for the group of Stratify 1 patients who tested positive for JCV antibodies.
- There was a dispute as to what the Skilled Team would take from the disclosure concerning Example 6. Dr Molyneux and Mr Baldwin considered that the Skilled Neurologist and Skilled IDS respectively would think that an nOD of 1.5 served to divide the anti-JCV antibody positive population in half and that it is that population split that the Skilled Team would consider significant. The key passages from their first reports are as follows:
"The Skilled Neurologist would be interested in the fact that 87% of samples from patients who developed PML fell within the upper half of all anti-JCV antibody positive index values obtained in the assay, whereas only 13% of PML patients samples fell within the lower half. Meanwhile, an index value of 1.5 separated the two halves of the antibody positive samples" [Molyneux 1, [164]].
"From the perspective of the [Skilled IDS], a key teaching is that the Gen1 assay (e.g. as described in Example 1 in the Patent and also, as the Patent explains, the subject of Gorelik and WO369) had served to divide the population into roughly 50:50 anti-JCV antibody positive and negative. However, the refined Gen2 assay in the Patent goes further, through the use of additional cut points, and serves to divide the anti-JCV antibody population into sub-groups as follows:
(a) ~20% low index value (<0.5) anti-JCV antibody positive
(b) ~30% mid-index value (0.5 to 1.5) anti-JCV antibody positive
(c) ~50% high index value (>1.5) anti-JCV antibody positive" [Baldwin 1, [157]].
- Neither Prof. Berger nor Mr Scrimshaw agreed that this is what the Skilled Team would understand the patentee to be disclosing as the significance of the 1.5 index value. Sandoz submitted as follows:
i) The hypothesis which the Patent advances is that risk of developing PML can be stratified by reference to anti-JCV antibody titer (expressed as nOD or index value). That is what the Patent then reports for both Gen1 in Example 4, and Gen2 in Example 6.
ii) Example 4 reports that there is an increased risk of PML above an nOD of 1.0 using the Gen1 assay on the basis that the rate of PML cases compared to non-PML cases is higher above an nOD of 1.0 than below it.
iii) Similarly, Example 6 reports that there is an increased risk of PML above an index of 1.5 using the Gen2 assay on the basis that the rate of PML cases compared to non-PML cases is higher above an index of 1.5 than below it.
iv) The Patent does not suggest that one can stratify risk of PML by dividing the population into fixed percentage groups, and such a suggestion would not make technical sense to the Skilled Team, because they would know that the proportion of seropositives varied depending on the population and would expect the same to apply to the distribution of index values amongst seropositive patients.
- In cross-examination, Dr Molyneux accepted that Example 6 does not define a clinical cut-off in terms of the proportion of the seropositive population that are above that cut-off. [T2 208/19 - 209/21]:
Q. Yes. But the clinical cut-point that they have defined is 1.5 in the Gen2 assay or the titer that corresponds to 1.5 in the Gen2 assay?
A. Yes, I agree.
Q. They have not said, "Your clinical cut-point is the 50% mark in any population". That is not what the teaching of the patent is, is it?
A. They have not made any statements about a different population. What they have said is that a clinical cut-point of 1.5 appears to strike a reasonable balance, as Professor Berger says, between those competing desires.
Q. That is because that is the titer -- it is that titer that is represented by 1.5 in the Gen2 population -- the Gen2 assay?
A. Yes, I agree.
Q. Do you agree that the Skilled Neurologist would not expect that the median anti-JCV antibody titer, amongst seropositives, would be the same in all populations?
A. I think the Skilled Neurologist would understand that it is possible that there would be differences in populations. The median value is unlikely to be precisely 1.50 in a different population.
Q. Right, okay.
A. And a consequence of that would be that a different proportion of patients in a different population, in fact different or significant, would fall above or below that cut-point. It would not be 50/50, it might be 45/55 or something similar to that.
- Similarly, under cross-examination at [T3 399/10 - 402/15] Mr Baldwin accepted that Example 6 was not saying that the clinical cut-off was represented by the median of the seropositive population; that if one were to use the median as the measure for whether a patient was at high risk or not, the answer would depend on the population being studied; that there was no reason to think that all populations would have the same distributions of titers within the seropositive population; and that therefore using the median will not identify the same group of patients at high risk (unless one assumes that the distributions of index values are the same).
- In his second report, Mr Scrimshaw had responded to Mr Baldwin's suggestion that the teaching of Example 6 was that the assay should divide the population into certain proportions, in this passage:
"I disagree that it is the teaching of EP 792 and specifically Example 6 and Figures 12 and 13 that the assay described in EP 792 should split the population 50:25:25 in the way Mr Baldwin describes. In effect, Mr Baldwin is saying that EP 792 defines 'high risk' as being the 25% of patients in a given population which have the highest anti-JCV antibody titers. However, this interpretation means that, if the average or distribution of anti-JCV antibody titers varies as between different populations, a different level of anti-JCV antibody titer will correspond to 'high risk' depending on the population tested.
Instead, the Skilled IDS would understand that EP 792 provides an index value (1.5) above which MS patients are viewed as being at high risk of PML when their serum is tested using the Gen2 assay. It is made clear that this index value is being used as a measure of or proxy for an underlying antibody titer above which risk increases (although this titer is not provided). As I explained at paragraph 228 of my First Report, an index value of 1.5 is only meaningful in terms of PML risk if the ELISA from which it is obtained provides the same index value output at the same level of anti-JCV antibodies as the Gen2 assay. If the ELISA differs from the Gen2 assay in terms of the assay set up or the cut-off calibrator then an index value of 1.5 no longer represents the anti-JCV antibody titer associated with a high risk of developing PML as described by EP 792.
The index value of 1.5 (and associated underlying antibody titer) was chosen by the inventors as being indicative of high risk on the basis that 87% of samples from PML patients fell above an index value of 1.5 compared to 50% of anti-JCV antibody positive patients - see paragraphs [0230]-[0231] - meaning that the risk of PML above that index value was higher than below it (where there were only 13% of PML samples and 50% of anti-JCV antibody positives). The Skilled IDS would understand that an index value of 1.5 in the Gen2 assay represents an underlying level of anti-JCV antibodies above which a patient has a higher risk of developing PML and that it had been arrived at by considering the ratio of PML samples to antibody positive patients above and below this threshold. They would not understand EP 792 to be teaching that what mattered was whether the index value was in the top 50% of the antibody-positive population (or the top 25% of the total population)."
- Although strongly challenged in cross-examination, Mr Scrimshaw's oral evidence was consistent with what he had said in his reports. He was clear in his view that the Skilled Team would understand that the Patent had set a threshold or clinical decision point at a particular titer or level of antibodies which corresponded to an index value of 1.5 in the Gen2 assay (but which would correspond to a different index value in a different assay).
- Similarly, Prof. Berger explained in his second report why he disagreed with Dr Molyneux on this point (and I agree with Sandoz that he was not effectively challenged on this evidence):
"As I have said, in paragraphs 164 and 198 of his report, Dr Molyneux seems to attribute significance to the fact that in Example 6 the index value of 1.5 divides the anti-JCV antibody positive population into halves. In my view, the purpose of the index value being set at 1.5 is not to achieve a division of the population in that way. Instead, the purpose of the index value is to represent an underlying antibody titer which is associated with a particular level of risk of developing PML.
Using the data from the particular patient population tested in Example 6, the patentee has sought to identify a threshold which groups the population into patients at higher and lower risk of developing PML. In determining that threshold, the patentee has identified an index value above which most (87%) of the pre-PML samples fell but that does not set the threshold so conservatively that the vast majority of anti-JCV antibody positive individuals who do not go on to develop PML are also deemed to be at high risk. The threshold has been set having regard to both (a) the proportions of pre-PML patients above and below the threshold and (b) the proportions of anti-JCV antibody positive but non pre-PML patients above and below the threshold.
The result of that analysis is that a particular index value (1.5) is identified. That particular index value (as recorded on the Gen2 assay) represents an underlying antibody titer. If the Gen2 assay were then used to assess risk of developing PML in another study population, that risk would be assessed depending on whether or not a particular individual had an index value of greater or lesser than 1.5. In this different population, the Skilled Neurologist would not expect an index value threshold of 1.5 to divide the seropositive population 50:50 nor that 87% of the pre-PML patients would fall above the 1.5 value. That is because the distribution of the anti-JCV antibody population above and below this index value would vary depending on the particular population tested. The Skilled Neurologist would be aware that seropositivity rates would vary depending on the population tested as I mentioned above at paragraph 6. They would also expect the spread of index values to vary across different populations, and would not expect that 50% of the antibody positive patients would have an index value > 1.5 in every population tested."
- The only cross-examination from Biogen which touched on the teaching of the Patent was at [T5 651/7 - 654/17]:
i) The first point that seemed to be being put to Prof. Berger was that the Skilled Team would appreciate that the teaching of the Patent was not just about the particular population of Example 6, but instead that Example 6 exemplified a relationship between antibody levels and risk which could be generalised to other populations. As Sandoz pointed out, that is entirely consistent with Prof. Berger's written evidence and their case.
ii) The second point that was being put was that the clinical cut-point could have been drawn at a different antibody titer. Again, that was consistent with Prof. Berger's written evidence and Sandoz's case. Sandoz submitted that the patentee could have chosen a different clinical cut-point, but it did not. At this point Sandoz submitted the patentee chose the antibody titer corresponding to 1.5 in the Gen2 assay.
- Example 7: This sets out two proposed statistical methodologies (Strategy-1 and Strategy-2) for assigning stratified risks to anti-JCV antibody positive MS patients based on their index value and % inhibition (as measured by the Gen2 assay in the STRATIFY-II study). Strategy-1 is described as a more conservative method, but the focus of Example 7 is on Strategy-2. Whilst the detailed statistics underlying Strategy-2 would be outside the expertise of the Skilled IDS and Skilled Neurologist, both would understand that the patentee proposes using index and % inhibition values to assign a patient to either a lower or higher risk group.
- Table 10 sets out, by reference to different index values for the lower risk clinical cut point, the empirical percentages of non-PML anti-JCV antibody positive patients that would be classified as lower risk if that cut point were adopted (see the second column) - so for example, if an index value of 1.5 was chosen as the cut point, 46.9% of patients would be assessed as lower risk. It also sets out the estimated percentages of future anti-JCV antibody positive patients that would be classified as lower risk if that cut point were adopted and the estimated percentages of future PML patients who would be misclassified as lower risk if that cut point was adopted. As the lower PML risk clinical cut point increases in value, the number of patients classified as lower risk increases but so too does the number of PML patients misclassified as lower risk, e.g.:
i) If the lower risk clinical cut point is 0.40, ≥ 9.2% of future patients would be assigned to the lower risk group and ≤ 1.2% of future PML patients would be misclassified as lower risk;
ii) If the lower risk clinical cut point is 1.25, ≥ 37.8% of future patients would be assigned to the lower risk group and ≤ 15.7% of future PML patients would be misclassified as lower risk.
iii) If the lower risk clinical cut point is 1.50, ≥ 43.6% of future patients would be assigned to the lower risk group and ≤ 22.2% of future PML patients would be misclassified as lower risk.
- Most attention was paid to the data for the cut-points at 1.25 and 1.50, so I will set out just that part of Table 10 (which sits just before the claims):
- Prof. Berger concluded from Fig. 12 and Table 10 that Biogen's selection of an index value of 1.5 in the Gen2 assay as the clinical cut off for lower risk patients represents a "reasonable balance between the risk of misclassification (which would decrease if the index threshold was lower) and the number of patients falling within the high-risk group (which would increase if the index threshold was lower). However, the Skilled Neurologist would appreciate that, based on the data in EP 792, a different threshold could have been selected by Biogen to differentiate between high and lower risk patients."
- In their written evidence, Dr Molyneux and Mr Baldwin made no comment about Example 7 and Table 10. In closing, Sandoz said that Example 7 and Table 10 show two points of significance:
i) First, they show that, whereas in the analysis done in Example 6 50% of 549 seropositive patients had an index value of < 1.5 in the Gen2 assay, when the population studied was increased slightly, to 595 seropositive patients, 46.9% of those patients had an index value of < 1.5 in the Gen2 assay. As Sandoz submitted, this shows that the chosen index value of 1.5 in the Gen2 assay does not need to divide the seropositive population into equal halves (a point eventually accepted by Dr Molyneux - see T3 213/10 - 215/19) and therefore confirms that one cannot use the median antibody titer amongst seropositives as some sort of surrogate for the antibody titer represented by an index value of 1.5 in the Gen2 assay.
ii) Second, Table 10 shows that the patentee has carefully considered various possible index values to use as the clinical cut-off, and the effect of choosing those different index values. It considers the effect of the choice of index value on, on the one hand, the percentage of patients who will not go on to develop PML who will be classified as lower risk and, on the other hand, the percentage of patients who will go on to develop PML having been misclassified as at lower risk. There is a balance to be drawn here, and what Table 10 shows is that, having considered various options, the patentee has decided to set the cut-off at an index value of 1.5 using the Gen2 assay - see Molyneux XX T2 215/20 - 217/8, concluding:
Q. What the patent decided to do here is to use the cut-off at an index value of 1.5 using the Gen2 assay; yes?
A. That is the claim of the patent as I read it, the cut-off above 1.5 defines the patient at high risk.
Q. Yes, and 1.5 using the Gen2 assay?
A. That is also correct.
Q. They have decided to go for that one rather than any other cut-point which they could have done, like 1.25?
A. Yes, Professor Berger makes a point in his report, and I agree, you could look at this and decide that a cut-off at 1.25 is also reasonable, but that does not detract from the fact that 1.5 is equally reasonable.
Q. And the one they have chosen is 1.5 for the Gen2 assay?
A. That is correct.
- Overall, Sandoz submitted that the evidence as to the teaching of the Patent is very clear and all one way. The teaching is that patients can be stratified into groups at high or lower risk of PML by reference to whether their antibody titer is above or below that represented by an index value of 1.5 using the Gen2 assay. It is that antibody titer which the patentee has, after careful consideration, decided to use as the clinical cut-point and it is that antibody titer which forms the basis of the method of the claims.
- Sandoz also submitted that Biogen's approach to the teaching of the Patent is driven by hindsight. Biogen pointed to the 2017 paper by Ho which discloses that (in the population of patients studied in that paper using the DxSelect assay), 87% of PML cases are to be found amongst the top 50% of seropositive patients. Biogen noted that this accords with the figures in Example 6 of the Patent. So, Biogen say, "we got it spot on". However, as Sandoz pointed out that population split corresponds to an index value of 1.7, not 1.5.
- Even allowing for that discrepancy, it is only with the benefit of the Ho data that Biogen can look back at the Patent and seek to build a case based on the fact that in Example 6 (but not Example 7) the median of the seropositive patients happens to correspond to an index value of 1.5 in the Gen2 assay. That information was not available to the Skilled Team and cannot be used to construe the claims or identify its teaching. The teaching of the Patent is that it is an antibody titer represented by an index value of 1.5 in the Gen2 assay that marks the clinical cut-off. Moreover, that index value is in the claims (which of course make no mention of any proportion of the population).
- In fact, Sandoz say that, far from showing that Biogen got it "spot on", the work done after the priority date shows that (as expected) the median of the seropositive population does not correlate with any fixed index value or antibody titer.
- The relevant legal principles of construction are well known and were summarised by Floyd LJ in Saab Seaeye Ltd v Atlas Elektronik [2017] EWCA Civ 2175 at [18]-[19], citing Virgin Atlantic v Premium Aircraft [2009] EWCA Civ 1062, [2010] RPC 8. Often it is enough to use this summary: the task is to undertake a 'normal' interpretation of the claims, which is an exercise in purposive construction. It is an objective exercise and the question is always what a skilled person would have understood the patentee to be using the words of the claim to mean.
- However, in light of the arguments on interpretation in this case, I consider it assists to set out the whole of the guidance from Saab Seaeye:
'18. There was no dispute about the principles which apply to the construction of patent claims. Both parties relied, as did the judge, on the summary in this court's judgment in Virgin Atlantic v Premium Aircraft Interiors [2010] RPC 8 ('Virgin') at [5]:
'(i) The first overarching principle is that contained in Article 69 of the European Patent Convention.
(ii) Article 69 says that the extent of protection is determined by the claims. It goes on to say that the description and drawings shall be used to interpret the claims. In short the claims are to be construed in context.
(iii) It follows that the claims are to be construed purposively - the inventor's purpose being ascertained from the description and drawings.
(iv) It further follows that the claims must not be construed as if they stood alone - the drawings and description only being used to resolve any ambiguity. Purpose is vital to the construction of claims.
(v) When ascertaining the inventor's purpose, it must be remembered that he may have several purposes depending on the level of generality of his invention. Typically, for instance, an inventor may have one, generally more than one, specific embodiment as well as a generalised concept. But there is no presumption that the patentee necessarily intended the widest possible meaning consistent with his purpose be given to the words that he used: purpose and meaning are different.
(vi) Thus purpose is not the be-all and end-all. One is still at the end of the day concerned with the meaning of the language used. Hence the other extreme of the Protocol - a mere guideline - is also ruled out by Article 69 itself. It is the terms of the claims which delineate the patentee's territory.
(vii) It follows that if the patentee has included what is obviously a deliberate limitation in his claims, it must have a meaning. One cannot disregard obviously intentional elements.
(viii) It also follows that where a patentee has used a word or phrase which, acontextually, might have a particular meaning (narrow or wide) it does not necessarily have that meaning in context.
(ix) It further follows that there is no general 'doctrine of equivalents.'
(x) On the other hand purposive construction can lead to the conclusion that a technically trivial or minor difference between an element of a claim and the corresponding element of the alleged infringement nonetheless falls within the meaning of the element when read purposively. This is not because there is a doctrine of equivalents: it is because that is the fair way to read the claim in context.
(xi) Finally purposive construction leads one to eschew the kind of meticulous verbal analysis which lawyers are too often tempted by their training to indulge.'
19. Sub-paragraph (ix) must now be read in the light of the Supreme Court's judgment in Actavis v Lilly [2017] UKSC 48, which explains that, at least when considering the scope of protection, there is now a second question, to be asked after the patent claim has been interpreted, which is designed to take account of equivalents.'
- Of course, the Actavis questions provide a way to identify differences between the claim and the alleged infringement which are immaterial to the issue of infringement. At the other end of the spectrum is the problem which arises when the claim is expressed using general or wide language. On that point, it is necessary to have regard to the well-known dictum of Floyd J. in Nokia v IPCom [2009] EWHC 3482 (Pat) at [41]:
'Where a patentee has used general language in a claim, but has described the invention by reference to a specific embodiment, it is not normally legitimate to write limitations into the claim corresponding to details of the specific embodiment, if the patentee has chosen not to do so. The specific embodiments are merely examples of what is claimed as the invention, and are often expressly, although superfluously, stated not to be "limiting". There is no general principle which requires the court to assume that the patentee intended to claim the most sophisticated embodiment of the invention. The skilled person understands that, in the claim, the patentee is stating the limits of the monopoly which it claims, not seeking to describe every detail of the manifold ways in which the invention may be put into effect.'
- Underlying that dictum is the important point made by Lord Diplock in Catnic Components Ltd v Hill & Smith Ltd [1982] RPC 183 at p242 that the wording of the specification and the claims is the choice of the patentee ('in words of his own choosing').
- The good sense of Floyd LJ's dictum is also reinforced by the fact that the Skilled Team has an understanding of patent law, although some care is required here as to how far one is entitled to go. In Kirin-Amgen Lord Hoffmann observed at [78] that the person skilled in the art must 'be assumed to know the basic principles of patentability'. This was applied by the Court of Appeal in Virgin at [15] per Jacob LJ:
'[15] We think it would unrealistic - indeed perverse - for the law to say that the notional skilled reader, probably with the benefit of skilled advice, would not know and take into account the explicit drafting conventions by which the patent and its claims were framed. Likewise when there is a reference to the patent being a divisional application, it would be perverse to work on the basis that the skilled man would not know what that means. A real skilled man reading a patent which, as in the case of the Patent, refers to "the parent application" would surely say "what's a parent application?" - and he would go on to ask a man who knows, probably a patent agent.'
- In the circumstances of this case, two parts of the Court of Appeal's reasoning have particular relevance:
i) First, the fact that the reader would in particular have in mind the fact that the nature of the two-part claim structure in which features found in the prior art are incorporated into the pre-characterising portion.
ii) Second, the Skilled Team would know about the practice of divisional applications and that this might affect their understanding of a claim because they will know that there are or may be aspects of what is described in the patent which are actually claimed in some other patent or patents divided out from the original application.
- Most issues of claim construction turn on the meaning to be ascribed to certain words or expressions in the claim. Part of the issue here is conventional in that there is an issue as to how '>1.5' should be interpreted, which I deal with below. The much more important and difficult issue relates to the breadth of this claim. I make no apology for discussing this latter issue at some length in this section of the judgment, because it is critical on virtually every aspect of this case.
- Although this section includes a number of references to technical evidence given by the expert witnesses, those parts of the evidence which I have accepted have educated me to understand what the words of the claim would be understood by the Skilled Team of the patent to mean. As usual, the issue of construction is one for the Court to decide.
- I repeat the wording of Claim 1 here:
"A method of evaluating a patient's risk of developing Progressive Multifocal Leukoencephalopathy (PML), the method comprising:
(i) determining, in a serum or plasma sample of the patient, an anti-JC Virus (JCV) antibody titer, wherein the anti-JCV antibody titer is determined by an ELISA assay comprising the following steps:
(a) forming a reaction mixture comprising an aliquot of sample and a substrate on which is disposed Highly Purified Viral-Like Particles (HPVLPs), and
(b) detecting the level of anti-JCV antibody bound to said substrate on which is disposed HPVLPs;
wherein the anti-JCV antibody titer is expressed as an index value, wherein the index value is determined by normalizing an optical density (OD) value of the sample to a cut-off calibrator adjusted to have an nOD of 1, and a positive control is adjusted to have an nOD of 1.3; wherein the cut-off calibrator and positive control comprise a mixture of serum positive for anti-JCV antibodies and serum negative for anti-JCV antibodies, and wherein a negative control comprises anti-JCV antibody negative serum and has an nOD of 0.1; and
(ii) determining the patient to be at high risk of developing PML if the anti-JCV antibody index value is determined to be > 1.5."
- With the teaching in the specification in mind, it can be seen that claim 1 includes some, but by no means all of the details of the Gen2 Assay. The following may be noted:
i) Although HPVLPs are a feature, they are neither limited nor approximated to the actual HPVLPs used in the Gen2 Assay. Nor is the make-up of the substrate.
ii) Certain features of how the index value is derived are taken from the Gen2 Assay, namely, that the cut-off calibrator is adjusted to have an nOD of 1, the positive control an nOD of 1.3 and the negative control an nOD of 0.1. However, it was clear that merely adopting or adjusting to those values does not provide the Skilled Team with the Gen2 Assay or an equivalent which would provide matching results.
- In other words, claim 1 claims a class of assays. I did not understand Biogen to dispute that. Indeed, Biogen strongly rejected Sandoz's suggestion that the claim was or had to be limited by the reading into the claim of the features of Example 6 where the clinical cut-off was 1.5. The evidence made it clear, in my view, that not every member of the class would be useful for the purpose of assessing the risk of developing PML. Indeed, in their closing, Biogen contended that Sandoz's real point was lack of utility, pointing out at the same time, as is the case, that lack of utility is no longer a ground of invalidity of a patent. Biogen also contended that the substance of the complaint was not correct because:
'The technical contribution is useful irrespective of where you chose to put the clinical cut-off. The Patent has taught an approach. The Patent has moved the art forward in a non-obvious way, which is the purpose of the patent system.'
- This quote exemplifies Biogen's whole approach, which was to focus on the teaching in the specification and largely ignore what the claim actually says. I say 'largely' because when in closing submissions I suggested to Dr Turner that the 1.5 figure was meaningless (because that cut-off represented a very wide range of possibilities, some of which would not be useful in assessing the risk of developing PML), his response was that 1.5 was not meaningless because, if you set your calibrator to 1, and your cut-off is 1.5, then you infringe, whereas if you set your calibrator so that your cut-off is 0.7, you don't infringe. This response rather proved my point. In essence, what Counsel was saying was that you can utilise the broad technical contribution of the Patent without infringing (assuming that some significance has to be attributed to > 1.5).
- Before proceeding further, I consider it useful to consider how Biogen's case developed on the interpretation of claim 1 and the related issues of the inventive concept or inventive core (used for the purposes of the equivalents issues) and the technical contribution. This development can be contrasted with the construction for which Sandoz argued, which remained more, but not wholly consistent.
- In their written closing submissions, Sandoz submitted as follows:
'Before the trial started, we had thought that, apart from a short point which goes only to infringement, there was no dispute about the construction of the claims. We had understood from Biogen's pleaded case and evidence that it agreed that the Skilled Team would understand that the index value of 1.5 and the antibody titer which it represents, above which a patient is determined to be at high risk of PML according to the claims, are those determined using the Gen2 assay of the Patent. We had understood that the only dispute was whether the Patent also taught that the Skilled Team could arrive at that clinical cut-off by identifying the upper 50% of seropositive patients.'
- Although I am well aware that it sometimes suits litigants to change (whether subtly or not) their characterisation of an important issue between opening and closing, in this instance, this paragraph accurately reflects Sandoz's Opening Skeleton Argument, which highlighted a passage from the evidence of Mr Baldwin which was entirely consistent with this view.
- Thus, Sandoz submitted as follows as to the interpretation of claim 1:
'132. We believe that it is common ground that the Skilled Team would understand that the index value of 1.5 in the claim and the anti-JCV antibody titer that it represents must equate to those determined using the Gen2 assay of the Patent and associated by the Patent with high risk of PML.'
- Biogen criticised Sandoz's construction on the following grounds:
i) That it relies on part of step (i) of the claim, namely '...determining, in a serum or plasma sample of the patient, an anti-JCV antibody titer...wherein the anti-JCV antibody titer is expressed as an index value...'.
ii) Accordingly, the 1.5 index value in step (ii) represents a titre of anti-JCV antibody.
iii) Therefore the 1.5 value in the claim represents the precise antibody titre that produced that index value in the Gen2 Assay, when it was used to generate the data shown in Figs 12 & 13.
iv) It therefore requires the precise reproduction of the Gen2 Assay.
- Biogen reminded me of the well-known dictum from Nokia v IPCom which I have already quoted. Biogen submitted that the 1.5 value in step (ii) of the claim cannot be sensibly read as importing every last detail of the Gen2 Assay into the claim.
- On that last point, Dr Turner accused Sandoz of writing insufficiency into the claim, which he characterised as wrong in principle. I return to this point later.
- As Sandoz pointed out in their closing, Biogen's written opening was notable for not addressing the construction of claim 1. Various statements, at differing levels of generality, were made about the teaching of the Patent or the technical contribution. At times Biogen suggested that what mattered was the fact that the clinical cut-point in Example 6 happened to be at the median of seropositive patients, while at other points they said that the technical contribution was much broader.
- The attempt to ride these two (or more) horses at the same time can be illustrated in Biogen's explanation of their positive case of sufficiency in their written opening (footnote refs removed):
'104. Biogen contends that the Patent is not about obtaining quantitative precision. At its broadest, the technical contribution of the Patent is the teaching - on the basis of sound data - that higher anti-JCV antibody titres are associated with a higher risk of succumbing to PML while receiving natalizumab (see in particular, Example 4, Example 6 and Table 10).
105. Example 6 (as illustrated by Figures 12 and 13) teaches how the patentee draws the line between patients at high and low risk based on anti-JCV antibody titre: the approximately 50% of antibody positive patients with the lowest titres are considered to have a low or lower risk of developing PML, and the approximately 50% with the highest titres to have a high risk (this latter group being associated with 87% of PML cases). This 'clinically relevant cut point' corresponds with an anti-JCV antibody index value of 1.5 as measured by the Gen2 assay. Thus, in the final analysis, index values > 1.5 corresponds with high risk of developing PML when the Gen2 assay is used (Figures 12 and 13).
106. However, the technical contribution of the Patent is not confined to the particular cut point adopted by the patentee to define high risk. Sandoz themselves point out that there is an element of subjectivity in choosing such a cut point (Amended Grounds of Invalidity, §5(c)). It is common ground between the clinicians that, with the benefit of the data presented in the Patent, different cut points could sensibly be drawn (see Berger 1, §134 and Molyneux 2, §35): see, for example, Table 10 which analyses the risk associated with other, more conservative cut offs.'
- Similarly, when it came to their equivalents case, Biogen submitted as follows:
'237. As explained above, Biogen contends that the broad technical contribution of the Patent is the teaching - on the basis of sound data - that higher anti-JCV antibody titres are associated with a higher risk of developing PML while receiving natalizumab (see in particular, Example 4, Example 6 and Table 10).
238. The claim is more narrowly drawn. Anti-JCV antibody titre must be determined by means of an ELISA assay that uses HPVLPs as the capture antigen. This must be expressed as an index value obtained by normalising to a cut-off calibrator made with a mix of anti-JCV antibody positive and negative serum. The patient is then determined to be at high risk of developing PML if that index value is > 1.5. This value derives from the teaching that, when the Gen2 assay is used, an index value of > 1.5 covers approximately the upper 50% of all anti-JCV antibody positive index values but 87% of PML cases (see Example 6).
239. While the claim requires anti-JCV antibody titres associated with a high risk of PML to be expressed as index values >1.5, the use of that index value as such to identify the cut point forms no part of the inventive concept of the claim. The same cut-point could be expressed by a different index value simply by adjusting the dilution of the calibrator. To use the technical contribution of the Patent it is sufficient to use an index value to represent a clinically relevant cut point to distinguish patients at greater risk of developing PML. That is the inventive concept or core of the claim.'
- In Biogen's oral opening, Dr Turner KC appeared to me to stress that the important point was the median of JCV antibody positive patients:
...........I am going
2 to come to insufficiency in a moment, but my Lord will have
3 picked up from our skeleton we say that if you want to put the
4 invention into effect, one way is to use an ELISA, use the
5 Gen2 ELISA, you do not have to, you can vary it or use an
6 alternative, and identify the 50% of JCV positive patients
7 with titers over the median and identify them as high risk,
8 and that is a straightforward thing to do.
- He reinforced the importance of the median by then going to a paper by Ho from 2017 where a much larger cohort of MS patients was analysed. He emphasised the congruence of the median (at 50.1% of JCV Antibody positive patients) at an index value of 1.7, which gave an estimated probability of PML of 87.3%, with the figures in Example 6, on the basis of which he submitted that this later study confirmed that the patentee, with the median in Example 6, was 'spot on'.
- Two points arise on Biogen's reliance on Ho. First, as Sandoz submitted, it indicates that Biogen's approach is driven by hindsight. Second, when I pointed out that the cut-off index value in Ho was 1.7 not 1.5, Dr Turner responded as follows:
13 MR. TURNER: What you look at is the percentage, not the cut off.
14 So with the cut off, you can set the cut off however you want.
15 What matters, the real thing, is if you go back to Figure 12,
16 if you take the median line, which is the 50/50, which is why
17 I focused on the 50/50, you can set your calibrator to call
18 that -- that could be 1.5, it could be 1.7, it could be 1.4,
19 it could be 1.3. It simply depends how you prepare your
20 calibrator. As my learned friend keeps emphasising, the
21 calibrator is not fixed against anything. Anyone can make up
22 a calibrator however they want. You need one for day-to-day
23 consistency, but it is not giving you a defined value.
24 However, what is a defined value is the median in Figure 12.
25 You could go 10% less than the median, 10% more than the
2 median....
6 So one needs to keep in mind that
7 you can choose how you set the calibrator, because the
8 calibrator is self-referential, but what matters is what the
9 antibody titer actually is, and the way you assess that is by
10 saying, "Is it the median of the population? (emphasis added)
- Slightly later, Dr Turner came to discuss the rival interpretations of claim 1:
'14 The question then arises is 1.5 a defined threshold?
15 This is where we both agree that an index of 1.5 is not of
16 itself a measure of antibody concentration, and depends on the
17 particular calibrator you are using. I think that much is
18 common ground. My learned friend says that means you are in
19 trouble! This then gives rise to two rival interpretations
20 which are before the court. One is that 1.5 is not, of
21 itself, setting a boundary in terms of antibody
22 concentrations, but in order to literally work the claim you
23 have to set your clinical cut-off at an index of 1.5. To be
24 precise, your clinical cut-off has to be an index of 1.45 to
25 1.54, because that is what 1.5 means. One might argue that
2 this is little more than a label, but it is none the worse for
3 that, particularly when the skilled person appreciates that
4 setting an index value is not, of itself, a measure of
5 antibody titer in the absence of international standards.
6 So that is our interpretation.
7 The other interpretation is that advanced by Sandoz at
8 paragraph 132 of their skeleton. They read off Figure 12 and
9 say that this represents a particular antibody titer which the
10 skilled addressee would understand to be unknowable from the
11 patent. So you know that the antibody titer is an unknowable,
12 but nevertheless 1.5 represents that unknowable titer, and
13 that unknowable titer is written into the claim as a
14 limitation.
- Reverting to Dr Turner's explanation of Biogen's construction, it seemed to characterise the index value of 1.5 as just an arbitrary number (as Sandoz submitted in closing), unless, it seems to me, the index values are derived from the Gen2 Assay or equivalent.
- In their written closing argument, Biogen characterised the rival constructions as follows:
'35. One of the central issues in the case (particularly for Sandoz's case of classical insufficiency) is one of construction: how is the index value of 1.5 in step (ii) of the claimed method to be interpreted?
36. Sandoz say that this value defines an absolute titre of anti-JCV antibody, namely that which was expressed as an index value of 1.5 when the patentee used the Gen2 assay to generate the data presented in Example 6 of the Patent. Biogen says that the value is not tied to any absolute titre of anti-JCV antibody, but is a means of expressing a dividing line between sero-positive patients at higher and lower risk of developing PML.'
- Dr Turner sought to support Biogen's construction with the following submissions:
i) That it offers a workable reading of the claim which is consistent with its language, the general teaching of the Patent, and the Patent's broad technical contribution as it would have been understood by the Skilled Team.
ii) Step (i) of the claim allows the Skilled Team to determine the patient's anti-JCV antibody titre using any appropriate ELISA that employs an HPVLP capture antigen, has controls and a calibrator as defined in step (i), and expresses that antibody titre as an index value. This freedom reflects the non-limiting wording of step (i) and the general teaching at [0011] - [0046] of the Patent.
iii) In step (ii) of the claim, the index value generated by that ELISA is used to determine whether the patient is at high or low risk of developing PML. The high-risk category is expressed by index values >1.5. However, that category can be associated with different ranges of anti-JCV antibody titres. This is because the cut off between the high and low risk categories is a choice to be made by the user in the light of the data provided by the Patent. The Patent does not dictate that the precise antibody titre corresponding with the clinical cut-off in Example 6 must be used to work the invention, and this would have been understood by the Skilled Team.
iv) To the Skilled IDS it is self-evident that the index value (nOD) in the claim is a relative value and that by its use the inventors are not seeking to communicate a particular antibody titre. As the court has seen, papers in this area (inter alia Gorelik) frequently cite nOD values which are not knowable as specific "titres" to the reader. The reason for this is that with polyclonal antibodies a true measurement of the quantity of relevant antibodies in a sample cannot be given but, more importantly, what is of interest to the reader is not the precise antibody concentrations being spoken about but the trends which are being identified and which can be reproduced by the reader.
v) Accordingly, Biogen's construction reflects the technical contribution of the Patent, as exemplified by Examples 4, 6 and Table 10. These Examples, for the first time, identify a correlation between higher anti-JCV antibody titres and the risk of developing PML, enabling stratification of the risk faced by the seropositive population.
- By reference to some cross-examination of Mr Scrimshaw, Biogen sought to bolster their construction with the following points, with which Mr Scrimshaw agreed:
i) In 2011 or 2012, the SAE, when presented with OD values quoted relative to a calibrator which was not available to the reader, would not see that as a communication of a particular analyte concentration because they would see it as a relative concentration, relative to the calibrator.
ii) The SAE reading Gorelik would not interpret it as teaching particular levels of antibodies because the calibrator and other details of the assay are not provided. They would only interpret it as teaching an approach which was useful but not directly repeatable. The SAE would be able to develop his or her own ELISA for detecting JCV antibodies.
iii) In the Patent, the SAE is not being told that the 1.5 index value relates to a specific level of antibodies because the patentee is not trying to communicate a specific titre. The value is only expressed relative to the patentee's calibrator and the patentee's assay set-up.
- For these reasons, Biogen said one had to 'park' the idea that one is dealing with an objective external measurement (examples being a given mass or temperature).
- It is worth spelling out how these points are said to support Biogen's construction. As I understand it, the argument involves the following steps:
i) The Skilled Team reads the Patent and understands that the cut-point index value of 1.5 is expressed relative to the calibrator, which is not provided.
ii) Therefore, the Skilled Team understand that the patentee is not and has not attempted to convey a specific antibody titre which corresponds to that index value.
iii) This is said to mean that Sandoz's construction cannot be correct and also that details of the Gen2 Assay cannot be imported into the claim.
iv) If Sandoz's construction cannot be correct, then (I assume the argument must be) the only other candidate is Biogen's construction, which reflects the technical contribution of the Patent.
- In closing, Biogen's case on construction was most clearly enunciated in these paragraphs where they summarised their case on classical sufficiency:
'82. Biogen's case is that to work the claim, it is not necessary to select the exact level of risk chosen by the patentee in Example 6 based on a precise antibody titre. The Patent teaches a broad technical contribution of PML risk increasing as antibody titre increases (see for example [0033] and [0039]) and anyone who is assessing risk by reference to antibody titre is using the technical contribution of the Patent. There has been no attempt to plead or prove that this relationship does not hold across a range of antibody titres.
83. To work the Patent, therefore, it is not necessary to read into the claim any particular threshold of risk. The term "high" is a relative term: those to the right of the chosen clinical cut-off being high risk. As we understand it, there is no dispute that in these circumstances the Patent is sufficient.
84. In the alternative, if the court is of the view that a limitation is to be read into the claim, as identified in Example 6 the skilled person can work the claim by applying the median value and identifying that by reference to another population.'
- The final point made by Biogen in their oral closing was that the 1.5 figure in the claim was to be viewed as the equivalent of sodium in the claim in the pemetrexed case - see Actavis UK Ltd v Eli Lilly and Company [2017] UKSC 48. It will be recalled that the claim in issue in that case claimed the use of pemetrexed disodium in the manufacture of a medicament (in combination with vitamin B12) for the treatment of cancer. The issue was whether pemetrexed dipotassium infringed. Having reformulated the Improver questions, the Supreme Court decided that it did because:
i) On the first question there was no doubt that the dipotassium version achieved substantially the same result in substantially the same way as the invention.
ii) On the second question, the skilled addressee of the Patent would appreciate that the dipotassium version would work in precisely the same way as the disodium version.
iii) On the third question, although the Court gave various reasons as to why the skilled addressee would not have concluded that strict compliance with the literal meaning of the claim was an essential requirement of the invention, I draw attention to two in particular: first, it was known that cations other than sodium could be successfully used with anti-folates and second, the skilled addressee would understand that the reason the claims were limited to the disodium salt was because that was the only pemetrexed salt on which the experiments described in the specification had been carried out.
- In the light of those considerations, Biogen's point is plainly bad. Potassium was a clear alternative cation to sodium which had no effect on the way the invention worked. In this case, although Table 10 sets out a range of possible alternative cut-offs which could be applied in the context of the Gen2 assay, none can be substituted for 1.5 because of the patentee's deliberate choice of 1.5. As soon as one departs from the specifics of the Gen2 assay, there could be a host of alternative cut-offs but there is no pointer to any of them, if the median argument has been dismissed.
- Drawing all these points together, Biogen's case on construction seemed to me to comprise the following group of concepts, from which a selection was made to address the particular point in question (whether construction, classical insufficiency, infringement etc). I list them in order of decreasing generality. To aid identification later, I have labelled each one, even though some are not feasible interpretations of the claim:
i) Construction 1: The 'broad technical contribution' that higher anti-JCV antibody titres are associated with a higher risk of developing PML while receiving natalizumab (e.g. Biogen Opening [104], [237], Closing [82])
ii) Construction 2: The 'inventive concept or core' of claim 1 is the use of an index value to represent a clinically relevant cut point to distinguish patients at greater risk of developing PML. This 'clinically relevant cut point' need not be at 1.5. (Biogen Opening [106] & [239], Closing [83]).
iii) Construction 3: As a variant of the 'inventive concept', the 1.5 value is not tied to any absolute titre of anti-JCV antibody, but is a means of expressing a dividing line between sero-positive patients at higher and lower risk of developing PML (Biogen Closing, [36], second sentence).
iv) Construction 4: The alternative 'median' case i.e. if the 'clinically relevant cut point' is at 1.5, this index value derives from the teaching (in Example 6) that 'the approximately 50% of antibody positive patients with the lowest titres are considered to have a low or lower risk of developing PML, and the approximately 50% with the highest titres to have a high risk (this latter group being associated with 87% of PML cases)' i.e. the median of antibody positive patients. (Biogen Opening [105] & [238], Closing [84]).
v) Construction 6: In the further alternative and more specifically, the index value of 1.5 as the clinically relevant cut point derives from the use of the Gen2 Assay. (see 'in the final analysis' in Biogen Opening [105] and the numerous references to Example 6), even though I understand that Biogen say this is not the correct construction. Biogen suggested that this was Sandoz's construction i.e. that 1.5 index value defines an absolute titre of anti-JCV antibody, namely that which was expressed as an index value of 1.5 when the patentee used the Gen2 assay to generate the data presented in Example 6 of the Patent (Biogen Closing [36], first sentence). Although in his oral closing, Mr Tappin KC denied that this was Sandoz's construction, it seems to me that at certain points in their argument, it clearly was. By way of examples: (1) see the suggestion made in cross-examination at [234.ii)] above; and (2) when addressing the numerical value issue (in the context of infringement), the clear submission was as follows (see [248] of Sandoz's written closing):
'On our construction the index value of 1.5 in the claim represents a specific anti-JCV antibody titer, namely that which equates with the titer determined using the Gen2 assay of the Patent and associated by the Patent with high risk of PML. Furthermore, on our construction this integer requires a patient to be determined to be at high risk if, and only if, their index value is > 1.5.'
- In closing, Mr Tappin KC was at pains to stress that Sandoz's construction did not involve reading into the claim all the details of the Gen2 Assay (i.e. their construction was not that summarised in v) above). Therefore, it is necessary to add this further possible construction: Construction 5: only those aspects of the Gen2 Assay which are explicitly mentioned in the claim are included. Mr Tappin distinguished between:
a) Those aspects of the Gen2 Assay which are mentioned in the claim e.g. a positive control with an nOD of 1.3, the negative control, and, so Sandoz submit, the cut-off of >1.5.
b) Those aspects of the Gen2 Assay which are not mentioned in the claim - the HPVLPs used, the dilution used etc.
On this basis, Sandoz submitted it is not permissible for Biogen simply to ignore the 1.5 limitation or treat it as meaningless.
- When considering Biogen's arguments, the following points occurred to me.
- First, as the opening words of step (i) indicate, step (i) in the claim is concerned with establishing the assay. In context, it is the assay by which the risk of PML is going to be measured. Step (ii) is the only part of the claim which addresses how the result(s) from the assay provide an evaluation of the risk of developing PML.
- Second, the points which Biogen say would be apparent to the Skilled Reader of the Patent must have been even more readily apparent to the Patentee. With those points in mind, it seems to me that any reasonable patentee (given the freedom to do so) would have expressed the claim differently, so as to avoid inclusion in the claim of a parameter with relative significance to a standard which was not specified. For example, although I am not going to formulate draft wording myself, in principle there does not appear to have been any conceptual or other difficulty in drafting a claim to reflect any of Constructions 1-3.
- Third, in the usual way, it is not productive to speculate as to the reasons why the patentee drafted this claim in the way that was done, or, similarly, as to the freedom or constraints on the patentee.
- Fourth, the advantage of stratification of risk of PML was very well understood in this art. See Gorelik and the two mentions of stratification in [0003] of the Patent where it is acknowledging the prior art. In very broad terms, the Skilled Team reading the Patent would understand the Patent was concerned with a further and new stratification of risk of PML, going beyond that in Gorelik and the other prior art mentioned in [0003].
- This expectation would be fulfilled on reading on in the specification. The key basic teaching is in [0033], [0034] and [0036] in passages which are worth repeating here:
'[0033] The methods disclosed herein are based at least in part on the discovery that anti-JCV antibody titer and other characteristics such as affinity/avidity can be indicators of a patient's risk of developing Progressive Multifocal Leukoencephalopathy (PML).
[0034] Accordingly, the invention features, a method of evaluating a patient's risk of developing PML, comprising acquiring knowledge of a JC Virus (JCV) antibody titer (e.g., determined as described herein and expressed as normalized optical density (nOD) or index) as defined in the claims.
[0036] If the titer or/and percent inhibition, or a function of both values is determined to be below a pre-determined level, the patient is determined to be at a lower risk of developing PML, and if the titer and/or percent inhibition, or a function of both values is determined to be at or above the pre-determined level the patient is determined to be at a higher risk of developing PML.'
- [0036] reflects Biogen's 'broad technical contribution' that higher anti-JCV antibody titres are associated with a higher risk of developing PML while receiving natalizumab.
- Taking the particular examples on which Biogen rely, Examples 4, 6 and 7 & Table 10, each teaches a dividing line (the 'pre-determined level') or dividing lines as follows:
i) Example 4, at an nOD of 1.0;
ii) Example 6, lowest risk, nOD <0.5, lower risk >0.5 nOD <1.5, higher risk nOD >1.5;
iii) Example 7 & Table 10, a series of individual dividing lines between lower and higher risk ranging from nOD values of 0.40 up to 1.50.
- So, the specification mentions or teaches a series of dividing lines. The Skilled Reader of the patent would note that, but would also note that the patentee has selected just one in claim 1, where the cut-off is at 1.5.
- The dividing line in claim 1 is expressed by reference to two relative terms: first, high risk vs low risk, and second, the parameter-less index value of 1.5. So, as Biogen pointed out, step (ii) includes two relative measures: high risk versus low risk and the > 1.5 index value.
- Although I have paid close attention to all the arguments presented to me and to aspects of the technical evidence, in my judgment the answer as to the proper construction of claim 1 is clear.
- First, it would be clear to the Skilled Reader that, when referring to the index value of 1.5, the patentee had deliberately not included all the details of the Gen2 assay. In my judgment, this claim is a paradigm for the application of Nokia v IPCom: any of the details of the Gen2 assay which are not set out in claim 1 cannot be read into the claim. By way of additional support, the Skilled Reader would understand that the patentee is likely to have claimed other aspects or combinations of features in divisional applications, so there is no need to read in features which are not specified in the claim.
- Second, I consider it is equally clear that the index value of 1.5 cannot be ignored or written out of the claim. That confirms that Biogen's 'broad technical contribution' argument (Construction 1) cannot be the correct interpretation of claim 1. If such a contribution were to be claimed, it would require very different wording. For essentially the same reason, that also excludes Biogen's 'inventive core' argument (Construction 2) precisely because an essential part of the argument is that the value of 1.5 does not matter. That index value of 1.5 is in the claim.
- Third, again for Nokia v IPCom reasons, it is impossible to read into claim 1 Biogen's 'median' construction (Construction 4). That would require reading into claim 1 the circumstances of Example 6, when the patentee did not write that into the claim. For the same reason, I can dismiss Construction 6.
- Fourth, in relation to the alternative inventive concept (Construction 3 - see [279.iii)] above), Sandoz suggested this was one of the most non-purposive constructions that could be imagined because it involves the following:
i) It doesn't matter whether the cut-off is at the titer which gives an index value of 1.5 in the Gen2 Assay.
ii) You can select whatever cut-off you like, but all that matters is that you set your calibrator so the answer comes out at the magic number of 1.5.
iii) In other words, 1.5 is just an arbitrary number.
- The key purpose of the claimed method is to distinguish between those at a higher risk of PML and those at a lower risk. The line of distinction is set by the cut-point of 1.5. Thus, the 1.5 index value is critical to the purpose. It must follow that a construction which means the 1.5 value is arbitrary is the very antithesis of a purposive construction.
- So, I have dismissed (with relative ease) each of the constructions for which Biogen contended. That does not mean that Sandoz's Construction 5 is automatically the right answer - it is not a question of the 'last man standing'. One point which I have pondered is whether Construction 5 means that the >1.5 cut-point in claim 1 is essentially meaningless. However, I consider it is clear that that cut-point is not necessarily meaningless: for example, on this construction, claim 1 includes the operation of that cut-point using the Gen2 Assay. This is a point I must return to when considering insufficiency.
- I have, however, concluded that Sandoz's construction is correct i.e. Construction 5.
- Accordingly, I find that the true construction of claim 1 is that it claims a class of assays which happen to employ the relative measure of the index value of 1.5 (and the other points specifically mentioned). It is, however, important to realise that claim 1 does not specify what that index value of 1.5 is relative to.
- In case I am wrong on construction (even though I do not consider I am), I will proceed to decide the remaining issues on the basis of Construction 5, but I will also state conclusions (as briefly as I can) on the basis of the other constructions which were argued. Construction 5 really only emerged in Sandoz's closing and the other constructions were in play throughout, so it is no surprise that submissions from each side on the issues which I have yet to decide were made on the basis of one or more of the other constructions, although sometimes it was not entirely clear which one was in contemplation.
- The issue here is whether the 1.5 value is a bookend in the claim, such that any value below 1.5 falls outside the claim, or whether 1.5 should be read as covering all values down to 1.45.
- Biogen, relying on Smith & Nephew v. Convatec [2015] RPC 32, submit that an index value of 1.45 or 1.451 (which they suggested was the level of precision received by the clinician) expressed to the same level of precision of the claim is 1.5. However, Smith & Nephew does not establish that 1.5 means 1.45-1.54, which is what Biogen submitted. It depends on the context.
- Sandoz submitted, in effect, that >1.5 meant exactly that. A patient is determined to be at high risk, if and only if, their index value is greater than 1.5.
- Sandoz pointed to the fact that the 1.5 cut point was determined using the data generated from Example 6, as illustrated in Fig 12. They point out that in Fig 12 the cut-point is indicated as the line (at 1.5) which divides high risk patients (to the right) from low risk patients (to the left). Fig 12 clearly shows there are points to the left of the 1.5 line, between 1.45 and 1.5, but those patients are characterised as low risk and not high risk. That is highly relevant context.
- There is a further practical point to consider. In practice, I have no doubt that if a clinician received an index value of between 1.45 and 1.5 for a patient, the clinician would have a serious discussion nonetheless about the risk of PML. That, however, does not affect the interpretation of this claim.
- In the context of this Patent, I agree that the index value of 1.5 represents a bookend to the claim. Thus, '>1.5' would be understood by the Skilled Reader to mean what it says: greater than 1.5.
- Finally, I should note that I received no argument about whether '>1.5' meant '
 ', but I would be inclined to find that it does. In my view, the Skilled Reader of the Patent would consider that, according to this method, a patient with an index value of 1.5 would be classified as high risk.
', but I would be inclined to find that it does. In my view, the Skilled Reader of the Patent would consider that, according to this method, a patient with an index value of 1.5 would be classified as high risk.
- For reasons which will become apparent, it is convenient to consider the insufficiency arguments first.
- Sandoz pleaded and pursued the following allegations of insufficiency:
i) Classical insufficiency, on the basis that the Skilled Team (and in particular the Skilled IDS) cannot, alternatively cannot without undue burden, produce an assay in which an index value of 1.5 represents the same anti-JCV antibody titer (and hence the same risk of PML) as it does in the Gen2 assay.
ii) Uncertainty insufficiency, because even if, by happenstance, the Skilled IDS does produce an assay whose results are the same as those of the Gen2 assay, the Patent does not explain how the Skilled IDS can determine whether their assay falls within the claim.
iii) A free-standing breadth of claim insufficiency attack against claims 1, 2, 3 and 7 which is based on the fact that the method of those claims does not work for patients who have previously been treated with immunosuppressants.
- It will be apparent that allegations i) and ii) were made on the basis of Construction 6, but allegation iii) is unaffected by the manoeuvring on the construction issue.
- In the course of argument, I raised a further possible ground of breadth of claim insufficiency if Construction 5 was correct. This was prompted by some of Biogen's submissions on the construction (and the breadth) of claim 1. Mr Tappin KC's response was that they had not considered that alternative claim, but that does not surprise me because of (a) Sandoz's understanding in the lead up to trial that there was only limited dispute over the breadth of claim 1 (see [257]-[263] above) and (b) the full range of the possible constructions in issue as to the interpretation of claim 1 only became apparent as the trial commenced and progressed.
- I propose to decide the pleaded allegations, having concluded that it is not necessary to address the further possible breadth of claim issue I identified.
- In relation to the three types of insufficiency, my attention was drawn to the following standard principles drawn from the authorities.
- The approach to classical insufficiency was summarised by Kitchin J in Eli Lilly v Human Genome Sciences [2008] RPC 29 at [239]:
"The specification must disclose the invention clearly and completely enough for it to be performed by a person skilled in the art. The key elements of this requirement which bear on the present case are these:
(i) the first step is to identify the invention and that is to be done by reading and construing the claims;
(ii) in the case of a product claim that means making or otherwise obtaining the product;
(iii) in the case of a process claim, it means working the process;
(iv) sufficiency of the disclosure must be assessed on the basis of the specification as a whole including the description and the claims;
(v) the disclosure is aimed at the skilled person who may use his common general knowledge to supplement the information contained in the specification;
(vi) the specification must be sufficient to allow the invention to be performed over the whole scope of the claim;
(vii) the specification must be sufficient to allow the invention to be so performed without undue burden."
- Claim 1 and the dependent claims said to have independent validity are each process claims. Therefore, the invention is identified as working the process (per statement iii) which must be capable of achievement without undue burden (per statement vii).
- The standard for undue burden was addressed by Aldous J (as he then was) in Mentor v Hollister [1991] FSR 557, 562 in the following terms which distinguish between 'prolonged research, enquiry or experiment' and 'ordinary trial and error':
"[The skilled person] must seek success. He should not be required to carry out any prolonged research, enquiry or experiment. He may need to carry out the ordinary methods of trial and error, which involve no inventive step and generally are necessary in applying the particular discovery to produce a practical result. In each case, it is a question of fact, depending on the nature of the invention, as to whether the steps needed to perform the invention are ordinary steps of trial and error which a skilled man would realise would be necessary and normal to produce a practical result."
- It has been said on many occasions that a patentee is "not entitled to set the reader of his specification a puzzle and call it a specification" (e.g. per Aldous J in Mentor v Hollister at p.563).
- In Evans Medical's Patent [1998] RPC 517 at pp536-537 Laddie J made the following observations regarding the instructions to be contained in the patent:
"It is not enough if the instructions are such that a number of equally qualified notional addressees can arrive at completely different end points, some within the scope of the claimed invention and some not. If reasonable addressees can come to different conclusions there is a conundrum as to which is right. That is not enablement. This view appears to be consistent with the approach of the Technical Board of Appeal of the EPO in Unilever/Stable bleaches (Decision T226/855) [1988] OJEPO 336, which was referred to with approval by Aldous J and the Court of Appeal in Mentor Corp v. Hollister Inc."
- The role of secondary evidence concerning insufficiency was discussed by Meade J in Gilead v NuCana [2023] EWHC 611 at [442]-[446], where he acknowledged its potential usefulness in giving "the possibility of a real-world cross-check on what the experts said was normal in this field".
- Uncertainty insufficiency was addressed by the Court of Appeal in Anan Kasei Co Ltd v Neo Chemicals & Oxides Ltd [2019] EWCA Civ 1646, distinguishing between a fuzzy boundary and conceptual ambiguity as follows:
"100. In the case of an invention which, ex hypothesi, is new it may not be easy to delineate the boundary with precision. In the same way as a conveyance of land may not tell you precisely where the boundary is, with the result that any dispute may have to be resolved by looking at topological features on the ground, so the boundaries of an invention may have to be determined as a matter of interpretation in the light of the common general knowledge that the skilled person would possess. But once that exercise has been carried out (these days including the possibility of equivalents), the court will be able to answer the question whether someone has crossed the boundary "yes" or "no". That, I think, is what Lord Hoffmann meant in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd [2004] UKHL 46, [2005] RPC 9 at [126] by a "fuzzy boundary" (a phrase which is now part of the jargon of patent lawyers). The boundary may be fuzzy, but it is still a boundary.
101. In my judgment [counsel for the Defendants] was right to submit that there is a difference between a fuzzy boundary in that sense, and a boundary whose location is impossible to ascertain. It may be impossible to ascertain because it is described in meaningless terms (the famous example of Pinocchio units given by Jacob J in Milliken Denmark AS v Walk Off Mats Ltd [1996] FSR 292); or because the patent does not explain how to decide where the boundary is (as in Kirin-Amgen itself). Patent lawyers have traditionally called this "ambiguity" but I do not think that that expression is accurate. Something is ambiguous when it is capable of having two (or more) meanings, and ultimately the court will be able to decide which of them is the correct meaning. Rather, in my judgment, the issue here is that of uncertainty. If the court cannot ascertain the boundary, having used all the interpretative tools at its disposal, it must conclude that the specification does not disclose the invention clearly enough and completely enough for it to be performed by a person skilled in the art."
- The example in Milliken was a "lie detector which had to be calibrated in Pinnochio units, no one knowing what these were". In Kirin-Amgen the claim in question was to recombinant EPO (rEPO) distinguished by having a higher molecular weight by SDS-PAGE than EPO from urinary sources (uEPO). But the patent did not identify the uEPO to be used as a comparator and the evidence established that not all uEPOs had the same molecular weight by SDS-PAGE, such that comparing a sample of rEPO with different samples of uEPO could give different results. The House of Lords held that in those circumstances the lack of clarity as to which uEPO to use meant that it was not possible for a skilled person to perform the invention.
- Sandoz also relied on Sandvik v Kennametal [2011] EWHC 3311, where the claim required a parameter to be within certain numerical limits (1.3 to 1.5). The patent required a comparison between measured intensity and a standard intensity, but did not identify the standard to be used (i.e. which PDF card to use) or whether to apply a particular approach (Kα2 stripping). The evidence was that the choice of PDF card, and whether or not Kα2 stripping was applied, each made a difference to the result (about 0.1 in the former case and about 5-10% in the latter case). Arnold J held that the patent was insufficient because it was uncertain what the correct test was to determine whether a product was within the claim.
- The Supreme Court considered breadth of claim insufficiency in Regeneron v Kymab [2020] UKSC 27 in the context of a product claim, setting out general principles in paragraph 56 of that judgment. Those principles were considered by Birss J (as he then was) in Illumina v Latvia MGI Tech [2021] EWHC 57 (Pat) and some were reformulated to cover process claims. The principles, some as revised in Illumina, may be stated as follows:
'i) The requirement of sufficiency imposed by article 83 of the EPC exists to ensure that the extent of the monopoly conferred by the patent corresponds with the extent of the contribution which it makes to the art.
ii) In the case of a product claim, the contribution to the art is the ability of the skilled person to make the product itself, rather than (if different) the invention.
iii) Patentees are free to choose how widely to frame the range of products for which they claim protection. But they need to ensure that they make no broader claim than is enabled by their disclosure.
iv) The disclosure required of the patentee is such as will, coupled with the common general knowledge existing as at the priority date, be sufficient to enable the skilled person to perform substantially all the types or embodiments [] within the scope of the claim. That is what, [], enablement means.
v) A claim which seeks to protect products or processes which cannot be performed by the skilled person using the disclosure in the patent will, subject to de minimis or wholly irrelevant exceptions, be bound to exceed the contribution to the art made by the patent, measured as it must be at the priority date.
vi) This does not mean that the patentee has to demonstrate in the disclosure that every embodiment within the scope of the claim has been tried, tested and proved to have been enabled [...]. Patentees may rely, if they can, upon a principle of general application if it would appear reasonably likely to enable the whole range [...] within the scope of the claim to be performed. But they take the risk, if challenged, that the supposed general principle will be proved at trial not in fact to enable a significant, relevant, part of the claimed range to be performed, as at the priority date.
vii) Nor will a claim which in substance passes the sufficiency test be defeated by dividing the [...] claim into a range denominated by some wholly irrelevant factor, such as the length of a mouse's tail. The requirement to show enablement across the whole scope of the claim applies only across a relevant range. Put broadly, the range will be relevant if it is denominated by reference to a variable which significantly affects the value or utility of the product or process in achieving the purpose for which it is to be performed.
viii) Enablement across the scope of a product claim is not established merely by showing that all products within the relevant range will, if and when they can be made, deliver the same general benefit intended to be generated by the invention, regardless how valuable and ground-breaking that invention may prove to be.'
- The parties were in agreement as to the target which the Skilled Team seeking to perform claim 1 had to meet. As Mr Baldwin explained:
'To put the invention into effect, the [Skilled IDS] needs to work up an anti-JCV antibody assay which identifies essentially the same population of anti-JCV antibody patients at high PML risk as described in the Patent, using the method described in claim 1.'
- He confirmed in cross-examination that by the "population of anti-JCV antibody patients at high PML risk as described in the Patent" he meant those with anti-JCV antibody titers above the threshold associated with high PML risk in Example 6, and that what the Skilled IDS needed to do was to devise an assay that could identify patients with antibody titers above that threshold and distinguish them from patients with antibody titers below that threshold.
- He also, in his first report described the task as being "to produce an assay which works in essentially the same way and has the same utility", which he explained meant an assay which would identify patients as being at high risk of PML if they would have been identified as at high risk of PML by the Gen2 assay described in the Patent, and being at lower risk of PML if they would have been identified as being at lower risk of PML by that assay, i.e. one where the boundary between high risk and lower risk is the same antibody titer as that which yielded an index value of 1.5 in the Gen2 assay in Example 6.
- Sandoz say the Skilled Team would be unable to reach the target because crucial information is missing from the Patent, in particular, (1) the identity and composition of the cut-off calibrator and (2) the identity and method of the production of the HPVLPs used as the capture antigen. Sandoz also say that, even if, by happenstance, the Skilled Team did manage to replicate the Gen2 assay, they would not know that they had done so - leading to uncertainty insufficiency. So this is a case where these two allegations of insufficiency have to be considered together, and it will be apparent where the uncertainty points arise.
- Mr Scrimshaw summarised these points as follows (emphasis added):
'227.1 The Skilled IDS would not be able to prepare the cut-off calibrator in EP 792 or an equivalent one. The identity of the cut-off calibrator is fundamental as all index values produced by the ELISA described in EP 792, including index values for patient samples, are measured relative to it. The cut-off calibrator is the fixed reference point of the ELISA in EP 792 against which all ODs are compared. The inability to reproduce the cut-off calibrator in EP 792 means that the Skilled IDS cannot develop an ELISA that provides equivalent results in terms of index values to the Gen2 assay in EP 792, in which the high-risk threshold of 1.5 was determined. The index value of 1.5 representing a high risk of PML in EP 792 will therefore not apply in any ELISA that the Skilled IDS produces. An index value of 1.5 in the ELISA produced by the Skilled IDS will represent a different anti-JCV antibody level, and therefore have a different clinical meaning and risk of developing PML, to an index value of 1.5 determined using the Gen2 ELISA described in EP 792.
227.2 EP 792 does not provide sufficient details regarding how to conduct an anti-JCV antibody ELISA of the type outlined in paragraph [0005] or the specific Gen2 ELISA that was used to determine the high-risk threshold of 1.5, including at least a failure to identify the capture antigen, certain reagents and assay protocol steps. The level of antibody binding and OD output of an ELISA is dependent on its set up, reagents and protocol and different samples will be affected differently by any variations in the ELISA due to the different polyclonal antibody populations in each sample. As a result, even disregarding the missing information about the cut-off calibrator, the Skilled IDS would not obtain equivalent nOD results to the Gen2 ELISA in EP 792. The cut-off threshold of an nOD of 1.5 for high risk of PML will not therefore also apply to any anti-JCV antibody ELISA that the Skilled IDS produces.'
- Reflecting their case on construction, Biogen made submissions at a number of different levels.
- In their Opening, Biogen started with a discussion of aspects of Sandoz's pleaded case, initially pointing out that they did not understand there to be any suggestion in Sandoz's expert evidence that the Skilled Team could not develop an ELISA assay for measuring anti-JCV antibodies and, when applying that assay to testing patient samples, obtain results which enable the Skilled Team to rank patients in terms of their relative JCV antibody titres (and, therefore, risk of developing PML). Nor, Biogen said, did it seem to be argued that the Skilled Team could not prepare a cut-off calibrator and assign it an nOD of 1, such that other antibody titres are compared to that reference point. The Skilled Team knows that the cut-off calibrator can be adjusted by selecting a sample with a high antibody concentration and diluting it to the desired concentration.
- Instead, the heart of Sandoz's complaint (as Biogen understood it) was that in seeking to follow the teaching of the Patent the Skilled Team's cut off calibrator may, in real as opposed to relative terms, be different to that used by the patentee and might therefore represent a different antibody concentration. In other words, the "clinically relevant cut-point" as represented in Figure 12 may fall further to the right or further to the left.
- Biogen drew attention to the following preliminary points which they said should be kept in mind when considering the allegations of insufficiency.
- First, Biogen acknowledged that there is no standard in the art against which to benchmark JCV antibody concentrations. The absence of a universal JCV antibody standard is the state of the art and is not a defect in the Patent.
- Second, OD measurements in an ELISA, in the absence of an external standard, are, to a greater or lesser extent, relative. The degree of colour change for any particular sample will depend on the way the assay is set up. Importantly, however, any competently prepared ELISAs will be appropriately controlled and able to measure and quantify the relative concentration of antibodies in samples, e.g. A>B>C>D.
- Third, adjusting and optimising assay conditions is the bread and butter of the skilled assay scientists. Alteration of assay conditions will usually happen across all samples; including controls and calibrators, including every time a new batch or a reagent is used.
- Fourth, in some fields greater precision is required than in others and so care needs to be taken in considering the context in which assertions of required accuracy are made. For example, if an ELISA is being used to measure the level of a particular reproductive hormone in the blood, it may be important to be precise in saying whether a hormone has reached a predetermined level - indicative of a reproductive state. If it is to be a commercial product, it would be required to perform consistently against a known standard: it will be necessary to ensure that different laboratories can achieve consistency of performance; that there is consistency between manufacturing batches; that the assay plates and reagents are stable over time and so forth.
- In another case, this level of precision may be unnecessary. An assay may be quantitative and require no particular quantitative precision: e.g. determining whether a patient has been exposed to Ebola or Covid virus by measuring the presence or absence of antibodies to that virus.
- Fifth, Biogen contended that the Patent in this case is not about obtaining precision against a known standard. The data in the Patent - inter alia because of the low number of PML cases available - is using a broad axe to distinguish cohorts with different risk profiles and in an otherwise featureless landscape this is extremely valuable.
- Then Biogen set out their positive case of sufficiency. I have already quoted the relevant paragraphs - see [264] above - and I have kept them in mind here.
- There is one key consequence. Biogen's principal case was that Sandoz's allegations of classical and uncertainty insufficiency were based on a misreading of the claim. Although Biogen did dispute certain of the allegations in Sandoz's case (such as the point about HPVLPs), in reality they were contending sufficiency at a higher level of generality, such as at Constructions 1, 2 or 3.
- With those introductory and relatively high-level points in mind, I can turn to consider the detail of Sandoz's allegations, starting with the cut-off calibrator.
- Biogen did not contend that the composition of the cut-off calibrator is provided by the Patent (and to that extent it agrees with Sandoz's case). Instead, in Mr Baldwin's first and second reports, two routes were put forward by which he said the Skilled IDS could reach the target and without undue burden. Each one requires assessment of some detailed facts. I discuss each of his routes in turn.
- In his first report, Mr Baldwin identified two pieces of information which he said would enable the Skilled Team to produce the required assay:
i) First, because the Patent "describes the key features of the Gen2 assay in Example 3" and
ii) Second because "it is the 50% of anti-JCV positive patients with the highest titres which correspond to those with an anti-JCV antibody index of greater than 1.5".
- It is necessary to examine his explanation as to how he said the Skilled Team would use those pieces of information to produce the necessary assay.
- In summary, Biogen relies on paragraph [0214] of the Patent which Mr Baldwin says identifies "key features of" or "key information" about the Gen2 assay by describing differences between the Gen2 assay and the Gen1 assay. Biogen also relies on Gorelik and/or WO369 for the sources of information about the Gen1 assay. In this way, Biogen proposes that the Skilled IDS could identify the reagents and protocols (or at least the main reagents and protocols) for use by the Skilled Team seeking to work the invention.
- Mr Baldwin explained that it would be necessary for the Skilled IDS to know the raw OD of the cut-off calibrator. However, he also explained that it would not be normal to define a cut-off calibrator by its OD, because it would vary from run to run. So, he said, "you cannot actually use the optical density to set the cut-off...you need to anchor it to a particular concentration". For this reason, Sandoz submitted that the whole exercise of trying to identify the cut-off calibrator for the Gen2 assay by reference to an OD is therefore flawed from the outset.
- The first point to note is that [0214] does not identify 'key features' of the Gen2 assay. It comprises a non-exhaustive list of differences between the Gen1 and Gen2 assays. Nonetheless, Mr Baldwin relied on the last bullet point of [0214] which I set out above but is worth repeating here. It reads as follows:
"The cut-off calibrator (CO) is adjusted to have a reactivity index of about nOD 1.0, and a positive control (PC) is adjusted to have a reactivity index of about nOD 1.3). The CO and PC are made by mixing an anti-JCV antibody positive serum and an anti-JCV antibody negative serum. For the negative control (NC), which is typically bottle negative sera, the reactivity index target is about 0.1;. Qualitatively, the controls come from different pools of human serum, but from an assay target concentration, they are similar to the Gen 1 control levels."
- Both sides were in agreement that the last sentence of this bullet point is not easy to understand. Mr Baldwin suggested that the Skilled IDS would understand the final sentence to mean that "quantitatively (i.e. from a target raw OD perspective)" the Gen2 controls were similar to those in the Gen1 assay, because "'assay target concentration' in this context must refer to the raw OD of the controls". He went on to say that the Skilled IDS would look up Gorelik and/or WO369 for information about the Gen1 assay and that, if they did so, those references would:
"confirm that the raw OD target for the cut off calibrator should be 1.0...and from that the raw OD of the negative and positive controls could be set at 0.1 and 1.3."
- Initially, Mr Baldwin said, after careful consideration, that the Skilled IDS would understand that the calibrator for the Gen2 assay should have an OD of 1.0 both if run in the Gen1 assay and if run in the Gen2 assay.
- Sandoz said there were five reasons why the Skilled Team would not read that final sentence in the way suggested by Mr Baldwin:
i) First, this sentence is not about the cut-off calibrator, but about the "controls". The Patent is, as can be seen from the 6th bullet point, distinguishing between the two and does not refer to the cut-off calibrator as a control.
ii) Second, even if one assumes that "controls" includes the cut-off calibrator in Gen2, Mr Baldwin's reading only succeeds in mapping the cut-off calibrator in Gen2 to the high positive control in Gen1. The final sentence of the 6th bullet point is clearly addressing the positive and negative controls in Gen2, yet on Mr Baldwin's reading it says nothing about the positive control in Gen2.
iii) Third, the sentence does not refer to raw OD, but to "assay target concentration" (emphasis added). But an OD is not a concentration - it is a unitless parameter. As Mr Scrimshaw explained, one possibility is that the reference to "concentration" may mean that the underlying level of antibodies in the controls was similar in the Gen1 and Gen2 assays, rather than that the ODs were similar. As he said, that would be logically coherent, though it was not clear whether that was what the Patent was trying to convey. Mr Baldwin's only answer was to seek refuge in "loose language" which he suggested in cross-examination was used by serologists (though such a proposition was never put to Mr Scrimshaw) and to suggest that there were examples where "concentration" was used as a substitute for "OD" or "nOD" (though no such examples were produced by Mr Baldwin or put to Mr Scrimshaw).
iv) Fourth, as Mr Scrimshaw explained, the Skilled IDS would expect differences between the Gen1 and Gen2 assays to have an impact on the read out of the assays in terms of raw OD. Therefore, a control with a raw OD of 1.0 in the Gen1 assay would not be expected to have a raw OD of 1.0 in the Gen2 assay. It would therefore be odd to compare the controls of the two assays in terms of their ODs. It was at this point that Mr Baldwin sought to withdraw his earlier answer that the Skilled IDS would understand that the calibrator would have an OD of 1 if run in either the Gen1 or the Gen2 assay. He also failed to come up with any coherent response to the point.
v) Fifth, it had been common ground between the experts in their reports that, when designing an assay that was intended to be run on a variety of spectrophotometers, the Skilled IDS would seek to ensure that the raw ODs generated by an assay fell within the dynamic range of most spectrophotometers (Mr Scrimshaw said that was 0.1 - 2.0/2.5, Mr Baldwin said 0.1 - 2.0). Further, Mr Baldwin agreed that because the Patent did not specify the spectrophotometer to be used, the assays it describes would have been designed to be run on a range of spectrophotometers. However, while use of the Gen1 assay generates Figure 11, with nODs up to 1.6, use of the Gen2 assay generates Figure 12, with nODs up to 4.2. If the raw OD of the calibrator of the Gen2 assay was 1.0, that would mean that the assay had been set up to generate raw ODs of up to 4.2. As Mr Scrimshaw explained:
"Such an approach would be contrary to one that the Skilled IDS would expect to have been taken based on their common general knowledge because an OD value of 4.2 is beyond the working range of most spectrophotometers. If the Gen2 assay had been designed so that the range of samples tested fell within the working range of around 0.1 - 2 or 2.5 OD, as the Skilled IDS would expect, then on considering Figure 12, the Skilled IDS would think it was likely that the Gen2 assay used a cut-off calibrator that generated a lower OD value."
- When this point was put to Mr Baldwin, he sought to depart from the evidence in his report. He suggested that whereas clinical chemists might adopt the approach he had indicated in his report, serologists would not mind ODs going up to 4. That was not a distinction he had drawn in his report, and indeed he had applied the same approach when he came to a serology assay in §164. Further, he suggested that all he had been saying was that one needed to have the calibrator and positive control within the dynamic range. That was not what his report said - indeed he had said in §164 that one needed to achieve OD values comfortably within the dynamic range for controls and samples.
- Two points were put to Mr Scrimshaw in cross-examination on this topic. First, it was put to him - on the basis of the Lee 2013 paper - that in fact the DxSelect assay used a calibrator with an OD of about 1, and so generated OD values of up to about 4. So, it was said, a real team did not follow the Skilled IDS's expectation. Sandoz accepted that that may be so and they suggested there may have been reasons for that, for example that Biogen knew that the spectrophotometer that would be used had a high dynamic range - no information was provided on that point. However, Sandoz submitted that has no bearing on what the Skilled IDS's expectation would be based on their CGK. Similarly, what GenBio did in 2019-2021 given the spectrophotometers then available to them has no bearing on the Skilled IDS's expectation in 2012. Indeed, Mr Andersen identified a specific reason why GenBio used the calibrator it did.
- The second point put to Mr Scrimshaw was that what mattered was whether the cut-off threshold was within the dynamic range. However, this ignores the fact that what Figure 12 shows is an assay which led to the determination of the cut-off threshold, which was previously undetermined. The assay could not have been set up with the cut-off threshold in mind (as GenBio could do in 2019-2021). Rather, the Skilled IDS would understand that the assay had been set up as described by the experts in their reports so that the results fell within the dynamic range.
- In his first report, Mr Baldwin had sought to support the derivation of a calibrator with an OD of 1 for the Gen2 assay from [0214] by saying that would have been in line with the Skilled IDS's expectation. However, in cross-examination he explained that people used anything from 0.2 up to 1.4 or 1.5 - "it is really horses for courses". When responding to Mr Baldwin's first report, Mr Scrimshaw was clear that a calibrator could have any OD within the working range of the spectrophotometer, and that it was neither standard nor a default option to choose a calibrator with an OD of 1. He was not challenged on that. The upshot is that the Skilled IDS's CGK does not lead them to an OD of 1 for the calibrator - Biogen must get that out of [0214] or not at all.
- In any event, Mr Baldwin proceeded on the basis that the calibrator in the Gen2 assay should have an OD of 1.0 (see the passage quoted in [348] above). But even if Mr Baldwin was right about the meaning of the final sentence of [0214], at its highest it says that the OD of the calibrator in the Gen2 assay is similar to that of the high positive control in the Gen1 assay. As Mr Baldwin said in cross-examination, "similar is not the measure that we can apply, is it?" Further, Gorelik and WO369 variously describe the OD of the high positive control in the Gen1 assay as "approximately" and "about" 1.0. As Mr Baldwin accepted, an OD which was "similar" to "about" 1.0 could be 0.9 or 1.1. Those would give different nODs for samples, and hence an nOD of 1.5 would give different clinical cut-offs. Mr Baldwin agreed with this example put to him in cross-examination: a sample with a raw OD of 1.5 would give an nOD of 1.36 with a calibrator with an OD of 1.1 and an nOD of 1.67 with a calibrator with an OD of 0.9.
Conclusion on this point
- Essentially for the reasons given by Sandoz, I find there was no sound basis for Mr Baldwin's assumption that the cut-off calibrator for the Gen2 assay would have an OD of 1.0, nor would the Skilled IDS come to that conclusion. On that basis, that puts an end to Mr Baldwin's first approach.
A postscript - what more could we have done?
- The final point to deal with on the topic of the cut-off calibrator is Biogen's plea: what more could we have done? As Sandoz suggested, just because a cut-off calibrator may be difficult to describe in words does not excuse a patentee from the normal consequences of failing to make an enabling disclosure of its claimed invention. However, Sandoz also put a solution to Mr Baldwin: Biogen could have made a batch of calibrator, stored it, and included a statement in the Patent saying that a sample was available on request. He said that would be burdensome for Biogen from a production/stability point of view, but, as Sandoz submitted, that is irrelevant - and if Biogen wants the benefit of a patent monopoly, it has to accept the burden of what is needed to make an enabling disclosure.
- It was put to Mr Scrimshaw that Biogen would have to provide a calibrator each time the assay was used. But that is hardly burdensome, as no doubt that is what Biogen actually does with its DxSelect assay. Further, it was not put to Mr Scrimshaw that if the Skilled Team had been supplied with a sample of calibrator by Biogen, they could not have used that to create their own calibrator (as Mr Baldwin had explained was possible). Mr Scrimshaw also explained that Biogen could have deposited a cell line expressing a monoclonal antibody and expressed their calibrator in terms of equivalents of the monoclonal.
- Finally, as Sandoz said, it is not for them to explain how Biogen could have made an enabling disclosure - it is up to Biogen to make an enabling disclosure if it wants a 20 year monopoly.
- The evidence given by Prof Roy is critical on this topic. However, and contrary to Biogen's arguments, I found aspects of Prof Roy's evidence difficult to follow.
- I can start with Prof Roy's agreement with §§71-73 of Dr Dugan's report, both as a matter of fact and as being CGK in 2012 [T3 266/21 - 267/13 & 268/7-14]. She therefore agreed to, inter alia, the following points:
i) VP1 can assemble into pentamers and under appropriate conditions into icosahedral capsid structures, though other structures can form depending on conditions. It is possible by purification to extract VLPs in the icosahedral capsid form, but the proportion of VLPs in that form will depend on the purification conditions. Generally antibody binding will be stronger to VLPs that more closely mimic the native capsid structure. The method of purification also impacts the level of impurities in the preparation; a low level of impurities is desirable to avoid antibody binding to those impurities.
ii) Most antibodies will be generated to epitopes on VP1, but antibodies will also be generated to epitopes on VP2 and VP3. The polyclonal mixture of antibodies generated will vary between individuals. The antibodies generated in any individual will also depend on the strain of JCV with which they were infected; the strength of binding between a VLP and antibodies in an individual sample will depend on whether the strain used to make the VLP is the same as that which infected the individual.
- The Patent makes it clear in [0119]-[0120] and in [0140]-[0144] that there are a number of options for the proteins used to make the HPVLPs and the degree of assembly of those proteins into larger structures. In particular, HPVLPs can be made entirely of VP1 or also include VP2 and/or VP3; different strains of JCV can be used; the HPVLPs can be in the form of a pentamer, or multiple pentamers, up to or in excess of the 72 found in the native capsid; the preparations can contain anything from 10 to 99% of HPVLPs in the form of the native capsid. Further different methods of purification can be used including one involving centrifugation and ultrafiltration, and one involving precipitation, concentration/diafiltration and ion-exchange chromatography. It is made clear that purity is important, not least because insufficiently purified VLPs result in high background noise yielding falsely high antibody levels.
- Prof. Roy accepted that the production and purification method could affect the way in which the resulting VLP preparation would perform in an assay, including for reasons given in Dr Dugan's report which she had accepted [T3 268/24 - 269/13]. She also agreed with Mr Scrimshaw that the identity of the HPVLPs was crucial as it determines the nature and level of binding to antibodies in the samples, that the particular HPVLPs used in the ELISA will affect the level of binding of antibodies in the sample to the immobilised HPVLPs, and that the way in which the HPVLPs are produced and purified will also affect antibody binding [T3 285/18 - 286/25].
- For these reasons, the nature of the HPVLP preparation was plainly a "key feature" of the Gen2 assay. In order to produce an assay which did what Mr Baldwin said it needed to do in §165 of his first report, it was plainly necessary for the Skilled Team to come up with the same HPVLP preparation.
- In order to achieve that aim, in §171 of his first report Mr Baldwin asserted that the HPVLPs used in the Gen2 assay were formed from VP1 and were the same as those used in the Gen1 assay. However, the Patent says neither of those things. It seemed that Mr Baldwin had simply assumed that, because the composition of the HPVLP preparation (as opposed to the coating concentration) is not mentioned in the six bullet points in [0214].
- However, as I pointed out above, [0214] starts by stating that the Gen2 assay differs from the Gen1 assay in "at least the follow [sic] ways". Mr Baldwin accepted that the list of six bullet points was clearly non-exhaustive, and so the Skilled IDS would have no reason to assume that there were no other differences between the assays, and would understand that there could be other differences that were not stated. Mr Scrimshaw's evidence was to the same effect. There is therefore no reason for the Skilled Team to assume that the HPVLP preparation used in the Gen2 assay was the same as that used in the Gen1 assay.
- Prof. Roy had said in §21 of her report that there was a "clear indication" in the Patent that the VLPs used in the Gen2 assay were the same as those used in the Gen1 assay. Her reasoning, in §20 of her report, appeared to be that because [0214] did not mention any differences, it would be assumed that there were none. That ignores the fact that [0214] is a non-exhaustive list, and is contrary to the evidence of Mr Baldwin and Mr Scrimshaw. In cross-examination, as Sandoz submitted, she was wholly unable to explain her reasoning in any coherent way (see [T3 280/4 - 285/10]).
- Sandoz went on to consider, contrary to their case so far, the position if the Skilled Team had concluded that there was a "clear indication" that the VLP preparation used in the Gen2 assay was the same as that used in the Gen1 assay. On that premise, Sandoz agreed that the Skilled Team would have looked at Gorelik and WO369 to see how the VLP preparation used in the Gen1 assay had been made.
- Sandoz accepted that both Gorelik and WO369 indicate that DNA encoding VP1 from the Mad-1 strain of JCV had been used to transform a baculovirus which was used to transfect SF9 insect cells. Gorelik cross-refers to the Sunyaev paper which sets out the purification process used. It is a purification process of the centrifugation / ultrafiltration type. The same process is set out in Example 1 of WO369. Example 7 sets out a different purification process, of the precipitation, concentration/diafiltration and ion-exchange chromatography type. Mr Baldwin accepted that, given that the aim was to replicate the VLPs used in the Gen1 assay, the Skilled Team would use the method that had been described in both Gorelik and WO369, namely the Example 1 method.
- Prof. Roy accepted that the physicochemical properties used to separate the materials in the Example 1 and Example 7 processes were different - in particular centrifugation separates on the basis of size and density, whereas ion-exchange chromatography separates on the basis of charge [T3 266/12-21]. She also accepted that outcomes could differ depending on the purification steps undertaken and in particular whether an ion-exchange step was included [T3 273/9 - 274/18, esp at 273/18-25 & 274/12-18]. And she stressed the importance of purity and of testing for purity [T3 274/3-6, 277/3-9, 278/10-14, 296/17 - 297/9]. Mr Scrimshaw also explained why purity of the HPVLP preparation was important, to avoid antibodies in the sample binding to non-JCV proteins on the plate leading to false readings (as [0144] says) [T4 544/5 - 546/18].
- Example 1 of WO369 does not contain any statement about the level of purity of the HPVLP preparation. Sunyaev simply includes an electron micrograph which shows that VLPs in the form of the native capsid have been formed. Example 7 states that its method "resulted in HPVLP preparations of about 80% HPVLPs" but an HPVLP according to WO369 is anything containing more than one pentamer - see p.6 l.28 - p.7 l.3. In these circumstances, Mr Scrimshaw said there is no reason to think that the two processes (Example 1 / Sunyaev and Example 7) would result in HPVLP preparations that behaved in the same way in an assay. Prof. Roy seemed to be suggesting at [T3 297/10 - 302/15] that in either case the Skilled Team would somehow end up with the same preparation consisting entirely of native capsid-like structures, but that appeared to be on the basis of applying further purification steps which were not stated in either Example of WO369.
- What this leads to is that the Skilled Team who thought there was a "clear indication" that the HPVLP preparation used in the Gen2 assay was the same as that used in the Gen1 assay would have followed the Sunyaev / Example 1 approach. However, it is clear that the DxSelect assay uses a different approach, which employs chromatography. That is apparent from the Lee poster [CXX/5] which states that whereas the Gen1 assay used "centrifugation-purified JC VLP", the DxSelect assay used "chromatography-purified and better-characterised JC VLP" [T3 383/7 - 384/7]. Further, the change to the "well-characterised" VLP is one of the factors to which Plavina [L/26] attributes the improved performance of the DxSelect assay [T3 386/16 - 387/23].
- Biogen has not revealed whether that change was already present in the Gen2 assay of the Patent. But, having declined to provide that information, Sandoz say that Biogen cannot assert to the Court that the Gen2 assay of the Patent in fact did use the same HPVLP preparation as the Gen1 assay, so that the Skilled Team who had followed the Sunyaev / Example 1 approach would in fact have ended up with the Gen2 assay HPLVP preparation.
- That is not the end of this point. Mr Scrimshaw pointed out that there were a number of other aspects of the Gen1 assay that were not disclosed even in the combination of Gorelik and WO369, including the particular detector antibody to be used (including the ratio to antibody to HRP and its commercial source), the buffers used to dilute the samples, detector antibody and capture antigen and wash the plates, and the volumes of each reagent to use. There was a dispute between the experts as to whether the Skilled IDS would assume that the Gen1 assay used the PBS-casein blocking buffer elsewhere in the assay - but I agree that Mr Scrimshaw's unchallenged evidence was more persuasive: Scrimshaw 2 §15, Baldwin XX T3 373/10 - 374/19.
- Mr Scrimshaw also explained that those factors could affect the output of the assay, and that they do not necessarily affect the calibrator and the samples equally. Mr Baldwin agreed - his point was that the differences were likely to be small [T3 321/6 - 322/4 & 372/6 - 373/9].
- Apart from the HPVLP preparations, there are further differences between the Gen1 assay and the DxSelect assay that are not disclosed by the Patent. The Lee paper [CXX/6] shows that the DxSelect assay employed MediSorp microtiter plates, with Focus' proprietary PBS buffer used as the coating buffer, and then blocked with Focus' proprietary blocking buffer. It also identifies the detector antibody that was used [T3 381/12 - 382/6]. Again, Plavina attributes improvements in the assay performance to, inter alia, the lyophilisation of the VLPs onto the plates, and the improved surface chemistry arising from the use of the MediSorp plates and the proprietary buffers [T3 386/16 - 387/23]. Mr Baldwin suggested that the differences between the various plates and buffers tested by Lee was small, but the data he relied on did not compare the various options with the various options the Skilled Team would need to select from when trying to implement the Gen1 assay approach, which did not involve lyophilising the VLPs onto plates at all [T3 391/21 - 393/19].
- Again, because of Biogen's failure to disclose the relevant information, we do not know whether the changes to the Gen1 assay that are present in DxSelect were also present in the Gen2 assay of the Patent. But again, given their position, Biogen cannot assert to the Court that these changes were absent from the Gen2 assay, such that it does not matter that they were not disclosed in the Patent.
- What we do know, as Mr Scrimshaw observed and as I pointed out above, is that the results from the Gen2 assay shown in Fig. 12 are very different from those from the Gen1 assay shown in Fig. 11. The most obvious difference is the change in scale of the nODs, but in addition the pattern of PML samples has changed. While there are some differences between the non-PML patient populations in Figure 11 and Figure 12 (It is unclear whether the populations were both drawn from Stratify 1 (as the figure legends suggest) and just differed in size, or whether Figure 11 comes from patients in the Affirm study - see Molyneux XX [T2 197/23 - 198/14]), the PML patient samples are likely to have been the same (as Prof Berger and Dr Molyneux agreed). So differences in the PML patient results must be because of differences in the assays, as Mr Scrimshaw said. But it remains unknown whether the changes shown and discussed in the Lee documents and in Plavina contributed to those differences in results.
- Sandoz identified two reasons why this matters. First, because the deficiencies in the disclosure of the Patent mean that even a Skilled Team who thought (wrongly) that the Patent was disclosing that the same HPVLP preparation was used for Gen2 as for Gen1 would still have choices to make (both between HPVLP purification processes and as to the other aspects mentioned in [376] above) which could affect the output of the assay and may well lead to them failing to produce an assay that was "functionally equivalent" (to use Mr Baldwin's term) to the Gen2 assay. Secondly, because even if by chance they had done so, they would not know that. Hence the need for Mr Baldwin's next step.
- Mr Baldwin recognised that the Skilled IDS who had produced an assay according to his approach would not know whether it corresponded to the Gen2 assay in the Patent and would therefore need to verify it. It was for that purpose that he employed what he said was the second important piece of information provided by the Patent.
- This was his point that the teaching of the Patent is that it is the top 50% of antibody positive patients that correspond to those with an index value of greater than 1.5. As Sandoz pointed out, that happened to be the case in the particular population of Example 6, but it was not the case in the slightly expanded population considered in Table 10. Further, as Sandoz submitted, the teaching of the Patent is that it is a particular antibody titer or level (namely that which gives an index value of 1.5 in the Gen2 assay) which marks the threshold between the high and lower risk groups. The Skilled Team would expect populations to differ in their distribution of antibody titers and hence index values, and would not expect that each population would have 50% of the seropositives above the threshold that the Patent has set. For this reason, I agree that the premise is flawed from the outset.
- Mr Baldwin explained how he said the Skilled Team would proceed in §183 of his first report. He said that the Skilled IDS would expect to match a distribution of ~50% seronegative, ~25% seropositive with index values < 1.5 and ~25% seropositive with index values > 1.5, and if it did not, would adjust the assay to match that distribution. That, he said, would lead to identification of "the higher risk, anti-JCV antibody positive population according to the teaching of the Patent", which he explained meant the same as "the same population of anti-JCV antibody positive patients at high PML risk as described in the Patent" in his §165 (i.e. in Example 6 - see [325] above).
- So it is clear that Mr Baldwin's approach was aimed squarely at producing an assay which would identify as high risk the group of patients who would be identified as high risk using the Gen2 assay of the Patent (and identifying as lower risk those who would be identified as lower risk using the Gen2 assay of the Patent), i.e. producing an assay with a cut-point at the antibody titer corresponding to an index value of 1.5 in the Gen2 assay. Sandoz agreed that is the right target (cf. [324]-[326] above). However, Sandoz went on to submit that the approach fails for the following reasons.
- In Mr Baldwin's third report, he said he sought to explain his reasoning in §183 of his first report in more detail. That involved pointing out that he had described the split between seropositives and seronegatives taught by the Patent as "roughly 50:50" and noting that he had used the ~ symbol when discussing the split amongst the seropositives. When asked about this at the start of his cross-examination, he was unable to explain further what he meant by "roughly 50:50" - the closest he came was to say that it might be 52/48 [T3 307/11 - 310/11].
- However, when he was later asked about §183, he made it clear that the split he said the Skilled IDS would be aiming for was 50/25/25, without any ranges around those figures [T3 395/11 - 396/5].
- The first problem with Mr Baldwin's approach is that he is aiming to produce an assay that identifies 50% of the population as seronegative. However, the proportion of seronegatives varies according to the population chosen. This can be established in various ways, (1) by looking at Table 2 of the Patent, which shows a range of seropositivity rates from 47.6% to 59.0% amongst the four groups studied (each of which contained between about 1000 and about 2500 patients). Similarly, (2) the McGuigan paper on which Dr Molyneux was a co-author [L/27] cited a study of 7700 patients showing variations in seropositivity rates between 48.8% and 69.5% across different European countries. (3) Variable results have also been found in other studies - see Berger 2 §23. Although Dr Molyneux suggested that some of the papers cited by Prof. Berger were considering patient populations that were not made up entirely of RRMS patients and might have given different results for that reason [T2 234/6 - 236/25], that point was not put to Prof. Berger. So a given population may well not contain 50% seropositives, yet Mr Baldwin's approach was striving to produce an assay that yields a seropositivity rate of 50%.
- Further, as Mr Scrimshaw pointed out in §65 of his second report, the assay that Mr Baldwin says the Skilled IDS would come up with is not capable of identifying those samples which would have been identified as seropositive by the Gen2 assay. As he explained, that is for two reasons:
"EP 792 provides nODs for these cut-offs as determined in the Gen2 assay (i.e. at 0.2 and 0.4) but the Skilled IDS would not know which nOD values from their screening assay would represent the same anti-JCV antibody titers as nODs of 0.2 and 0.4 from the Gen2 assay. Further, the Skilled IDS would need to use a confirmatory assay to assign any indeterminate samples as positive or negative. The Skilled IDS could set up a confirmatory assay but they would not know whether their confirmatory assay would produce 45% inhibition at the same point as the Gen2 assay and therefore whether their confirmatory assay would assign indeterminate samples as positive or negative in the same way as the confirmatory step of the Gen2 assay in EP 792."
- Mr Baldwin was unable to deal with this point. He suggested that one would get an idea of which samples were negative by looking to see which had an absorbance similar to that of the assay buffer, but that would obviously not pick up all those samples which would be identified as negative by the Gen2 assay [T4 415/7 - 417/23]. Then he suggested that one would be able to identify the cut points for seronegatives that equated to index values of 0.2 and 0.4 in the Gen2 assay. But that was "later on" once the 1.5 cut-point had been identified, so the whole process was circular - in order to split the seropositives 50/50 to find (on Mr Baldwin's approach) the antibody titer which corresponds to 1.5 in the Gen2 assay you first need to be able to identify the seronegatives. It was at this point that Mr Baldwin said, completely aptly, that it was "like trying to build a house on quicksand".
- Mr Scrimshaw was not actually challenged on §65 of his second report. All that was put to him was that one could use "the Gorelik approach" to determine which samples were seropositive and which were seronegative. This could be tried, but the problem identified by Mr Scrimshaw in §65 in his second report remains.
- Even if somehow the Skilled Team were able to identify the seronegatives, leaving only the seropositives to be split 50/50, as Mr Baldwin suggests, the Skilled Team would still end up with the wrong answer (or even if by chance they ended up with the right answer, they would not know that).
- Here the problem is that index value distributions (and hence distributions of the underlying antibody titers) vary between populations. That can be seen from a number of studies all of which used the DxSelect assay. When considering these, it must be kept in mind that, while Mr Baldwin said that the Skilled Team would use samples randomly selected from the general population, Biogen's evidence was that there was no evidence that index values in healthy volunteers differed from those in MS patients.
- Plavina [L/26] studied a number of populations using the DxSelect assay:
i) A group of 100 healthy volunteers gave a distribution of index values with the 25th percentile at 0.25, the median at 1.04 and the 75th percentile at 2.4. Biogen has not revealed what the proportion of seronegative individuals was, but there is no suggestion that the median in the seropositive population was 1.5.
ii) The test data set (made up of 1039 non-PML patients from the Affirm and Stratify-1 studies plus 45 PML patients) showed a median amongst seropositive patients of 1.4.
iii) The verification data set (made up of 1,483 non-PML patients from the Stratify-2 study plus 26 PML patients) shows a median amongst seropositive patients of 1.9.
- Ho [L/28] studied an index cohort made up of 8170 patients from Affirm, Stratify-1 and (mostly) Stratify-2 using the DxSelect assay. They showed that the median index value amongst seropositive patients was 1.7. (As mentioned above, Biogen now appears to like these data, because the median index value was also associated with 87% PML cases. But that is irrelevant when the Skilled Team is trying to identify the antibody titer associated with an index value of 1.5 using the Gen2 assay and is faced with varying populations.)
- Prof. Berger also referred in §24 of his second report to further studies, including a study he had been involved in using DxSelect in a group of Austrian MS patients, where the median index value among seropositive patients was 2.3, and a study by Bonek using DxSelect in Polish MS patients which reported a median index value among seropositive patients of 2.2 (and which referred to a variety of other median index values amongst seropositive patients in other studies).
- Because the distribution of index values, and hence underlying antibody titers, varies amongst seropositive individuals and varies depending on the populations studied, the Skilled Team which tried to adopt Mr Baldwin's approach of identifying the cut-off which corresponds to an index value of 1.5 in the Gen2 assay by dividing the seropositive population 50/50 would be likely to come up with the wrong answer, and even if by chance they came up with the right answer they would not know that they had done so.
- Therefore, Mr Baldwin's approach would not enable the Skilled Team to identify the same population of seropositive patients at high risk of PML as described in the Patent, which was the objective (see [325] above).
- Mr Baldwin's attempt to address that problem seemed to involve trying to replicate the pattern shown in Figure 13 of the Patent. However, it was very unclear how he said the Skilled Team would be able to achieve that, because he accepted that the distribution of results one got would depend on the population chosen. This seemed to involve carrying on testing until one found a pattern that looked like Figure 13, at a high level of granularity, but he was unable to explain how the Skilled Team would manage to achieve that [T3 399/3 - 402/15 & T4 426/25 - 435/9]. In any event, the fact that a pattern looks like Figure 13 does not mean that the median of the seropositives is at the same antibody titer as that corresponding to 1.5 in the Gen2 assay. Mr Baldwin's suggestion of trying to match Figure 13 of the Patent to identify the cut-off threshold of the Patent was not put to Mr Scrimshaw.
- Biogen's refrain was that its DxSelect assay works in all populations, and I think the suggestion being made was that differences between populations do not matter for the purpose of this case. Sandoz submitted that is looking at things through the wrong end of the telescope, for the following reasons. The DxSelect assay can be used in all populations because it identifies a certain antibody titer (represented by an index value of 1.5, although (as already pointed out) we do not know if that is the same antibody titer that corresponds to an index value of 1.5 in the Gen2 assay). For that purpose it does not matter what the distribution of index values is in any given population. If the median index value amongst seropositive patients is higher in a given population, the outcome is simply that a higher proportion of that population will be classified as high risk (because a higher proportion will have index values > 1.5).
- As Sandoz pointed out, the issue facing the Skilled Team on Mr Baldwin's approach is a different one, namely trying to identify the antibody titer which corresponds to 1.5 in the Gen2 assay by identifying the median amongst seropositive patients in a population which may differ in terms of antibody titer distribution from that used in Example 6 of the Patent.
- It appeared from the cross-examination of Prof. Berger that Biogen were trying to establish a different point: namely that the Skilled Team seeking to identify the antibody titer that marks the clinical cut-off of the Patent would use a population that was matched for age, gender and geographical location with the Stratify-1 study (that used in Example 6), and then look for the median amongst the seropositives in that population. That was not an idea which was advanced by Dr Molyneux, Mr Baldwin or any other expert. In any event, there is no evidence to support the suggestion that this would solve the problem. Sandoz submitted that, in fact the information we have indicates that it would not, for the following reasons.
- First, the same problem of identifying the seronegatives, discussed above, would remain.
- Secondly, as seen above, while in Example 6 an index value of 1.5 split the seropositive population 50/50 when 549 seropositive patients from the Stratify-1 study were examined, when a further 46 patients from that study were included, an index value of 1.5 split the seropositive population 46.9% / 53.1% as shown in Table 10. That shows that even including a few more patients from a study with a given age/gender/geography profile can give a different answer.
- Thirdly, the age/gender/geography profile of the Stratify-1 study was almost identical to that of the Stratify-2 study, as can be seen from looking at the data in Table 2 of the Patent (Stratify-1) and in Table 1 of Ho (Stratify-2). Both were conducted solely in the US. In Stratify-1 the mean age was 44.4 and the median age was 45, while in Stratify-2 the mean age was 44.1 and the median age was 44. In Stratify-1 the gender split was 75.7% female / 24.3% male, while in Stratify-2 it was 74% female / 26% male.
- Yet when Plavina [L/26] studied the Stratify-2 population (in the verification data set) the median index value amongst seropositive patients was 1.9. This again shows that even with matched age, gender and geography, different populations give different median index values amongst seropositive patients.
- In §§13-18 of his second report, Mr Baldwin introduced a new approach, which sought to avoid the issues around [0214] of the Patent. Sandoz said they were unable to detect Mr Baldwin's second approach being put to Mr Scrimshaw, and they assumed it has been abandoned, but I can deal with it relatively briefly.
- Mr Baldwin's aim was to make what he called a "functionally comparable ELISA", which he confirmed meant one which produced the clinical threshold which is equivalent in terms of antibody titer and level to the index value of 1.5 in the Gen2 assay of the Patent. So the target was the same as with his first approach - it was the means of trying to get there that was different.
- Initially Mr Baldwin resisted the suggestion that the second approach was different to the first, but then conceded that it was. When asked why, if it was an approach that he said would have occurred to a Skilled IDS, it was not in his first report, he had no convincing answer. He suggested that he had wanted to save space in his first report, but his second approach only occupied five paragraphs in his second report. I agree that the inference is clear - it was one that had not occurred to him when he wrote his first report.
- In any event, the second approach ran into the same problems as the first. The idea was to run out 200 samples from the general population, and then create a calibrator so that normalising against its raw OD "results in the clinical threshold shown in Figure 12/13 occurring at an nOD of 1.5" (see §17). The "clinical threshold" was said to be disclosed in the Patent (see §18). When Mr Baldwin was asked what he meant by the clinical threshold disclosed in the Patent and shown in Figures 12/13, he said that it was the antibody titer or level that corresponds to an index value of 1.5 in the Gen2 assay. However, as he accepted, in order to identify that threshold, he had to apply his 50/25/25 split and try to reproduce Figure 13, so the same problems arise as with his first report.
- What was put to Mr Scrimshaw appeared to be that the Skilled Team could identify the median amongst seropositives in a given population, and then choose a calibrator such that that median gave a value of 1.5 (or even so that a value of 1.5 represented some other point in the distribution of seropositives, e.g. 40% or less). Of course, the problem about identifying the seronegatives, so that the seropositives could be divided 50/50, would remain. Mr Scrimshaw did not accept that this is what the Skilled Team seeking to put the Patent into effect would do. On the contrary, as he pointed out, Biogen's approach would mean that the clinical threshold that would be identified was not the same as the one defined (and claimed) by the Patent [T4 503/11 - 508/8].
- This approach was put to Prof. Berger in cross-examination [T5 655/18 - 659/19] in a somewhat unusual fashion. First, counsel said that he wanted to consider how the skilled person could use the information in the Patent (not how the skilled person would use the information if they were trying to use the claimed invention). Then various things that "you could" do were put to Prof. Berger (including an assumption that "you like the risk profile...that divides the population 50:50"), without making it clear what he was being asked to agree was "an approach which could be taken". Then it was put to him, on the basis of Ho, what you would have found if you had looked for the median.
- The final proposition put (with the data in Ho open) was that "let us assume you picked a population where the median was not quite in the same place. Let us say you had gone for a 50 - let us look at the 1.5, you have 55%, so you picked a population and you have ended up picking that 55% in one side and 45% of the other. You would have 90% of your PML patients to the right of your line."
- Sandoz were of the view that this was probably intended to be the point also put to Mr Scrimshaw that one could have chosen to divide the population not 50/50 but, say, 55/45.
- When counsel returned to the topic after questions from me, Sandoz submitted he started with a long and complex "question" which was a mix of statements about Biogen's case and assumptions about putting "the general teaching of the patent into effect". The final question was about coming up with some "prototype assay - I am not talking about natalizumab - which has some potential use"; it was unclear what counsel was hoping to get at here.
- I agree that the cross-examination of Mr Scrimshaw and Prof. Berger wholly failed to establish that the Skilled Team seeking to implement the teaching of the Patent would have proceeded in the way suggested by counsel for Biogen. As Sandoz pointed out, his approach was not one supported by Biogen's experts in any event.
- In closing, Biogen made a number of submissions by which they attempted to deflect the force of Sandoz's case. These were founded on their contention that Mr Scrimshaw accepted that the Skilled IDS would have been able to 'work the ELISA described by the claim'. Biogen relied on two passages of his cross-examination.
- First, Biogen said Mr Scrimshaw accepted that there was no difficulty in setting the calibrator to correspond to any particular clinical cut point:
11 Q. So that is the point. I am going to come back to calibrators,
12 but I understand you can set the calibrator wherever you want,
13 as I understand?
14 A. Yes.
- Second, Biogen contended he was taken through the claim and accepted it could be performed, relying on the following answers:
8 Q. Just following through, they could determine the serum or
9 plasma sample of the patient and identify an antibody titer
10 wherein the antibody titer is determined by an ELISA? They
11 could do that; yes? We discussed that?
12 A. Yes.
13 Q. They could use the HPVLPs; yes?
14 A. Yes.
15 Q. And detect the level of antibody bound to the substrate?
16 A. Yes.
17 Q. And they could express the anti-JCV antibody titer as an index
18 value?
19 A. Correct.
20 Q. And they can adjust -- I was going to say the cut-off
21 calibrator will be an nOD of 1.
22 A. Yes, it is not adjusted. Just by definition it is going to be
23 1.
24 Q. They could set their positive control at 1.3; yes?
25 A. Yes.
2 Q. And they could also have a negative control, that would be
3 routine; yes?
4 A. Yes.
5 Q. Then they could determine the antibody level of the patients
6 and they could choose their cut-off calibrator, their clinical
7 cut-off, and then set their calibrator to 1 such that their
8 index value was 1.5?
9 A. Yes.
10 Q. There is no problem about performing claim 1?
11 A. The problem is, in my mind, in my opinion, that that level of
12 1.5 in a new assay is not going to represent the same titer as
13 in the original one, as detailed.
14 Q. Right. Why does it have to?
15 A. Because that is your clinical threshold for higher risk.
- Biogen characterised his only objection in this way: if you set the cut off by reference to a different population, it may not represent the exact same titre as in Example 6 of the Patent. This submission was entirely consistent with Biogen's case seeking a construction at an impermissibly high level of generality - the 'broad technical contribution' (Construction 1). On this basis, Biogen made a number of submissions to the effect that any difference(s) would be de minimis and/or if there were differences, they did not mean that the teaching of the Patent was not useful. I did not find any of those submissions persuasive, all being pitched at the wrong level of generality.
- In their opening skeleton, Sandoz explained their secondary evidence of insufficiency, founded on what GenBio had to do to come up with the Sandoz Assay.
- As I mentioned, Dr Andersen explained the development of the Sandoz Assay in his witness statement. The following summary is based on [191]-[192] of Sandoz's opening skeleton but I have omitted two pieces of confidential information which concern sub-paragraph iv) below (which I nonetheless noted):
i) that GenBio's instructions were to develop an assay which produced results as close as possible to those of Biogen's STRATIFY JCV DxSelect assay;
ii) that GenBio did not have access to the STRATIFY JCV DxSelect assay, but did have access to serum samples which had been tested using that assay, together with the results of such testing. That meant that GenBio could compare the results from the assay it was developing with those obtained using the STRATIFY JCV DxSelect assay and adjust the design of its assay to seek to match the results being produced by the STRATIFY JCV DxSelect assay;
iii) that GenBio tested six JCV VP1 antigens from five different sources, of which only two showed promise; the antigen concentrations of these two were then optimised and one antigen was selected for further development;
iv) how GenBio went about adjusting the parameters of the assay so that it produced results similar to those generated by the STRATIFY JCV DxSelect assay and how it developed a suitable calibrator and controls;
v) that the development work using the initially selected antigen took over a year;
vi) that it became necessary to change the antigen and use the back-up antigen instead and to repeat the development work, which took another 4-5 months.
- The process undertaken by GenBio (and in particular ii) and iv) above) was summarised in Scrimshaw 1 §252:
"Paragraph 12 of Andersen explains that GenBio had access to a range of serum samples from Polpharma's clinical trials for which it had index value results from the STRATIFY JCV DxSelect ELISA. GenBio then used these serum samples and their respective index values as determined by the STRATIFY JCV DxSelect ELISA to adjust the anti-JCV antibody assay they had developed to align the output of their assay to the STRATIFY JCV DxSelect ELISA. In the circumstances described in Andersen, in which GenBio had no access to the STRATIFY JCV DxSelect ELISA itself, this iterative process of comparing results and revising the assay was the only way in which GenBio could have developed an ELISA that provided index values that correlated with the STRATIFY JCV DxSelect ELISA."
- Accordingly, as Sandoz submitted, their assay was prepared only through Sandoz's ability to obtain results from Biogen's STRATIFY JCV DxSelect assay for samples it could also test on its own assay - i.e., information which is not disclosed in the Patent. Even with access to results from the STRATIFY JCV DxSelect assay, which the Skilled Team would not have had, it took about 18 months for GenBio (after another company had failed) to produce an assay that corresponded as well as possible to STRATIFY JCV DxSelect (a point relevant to infringement which I consider below). Sandoz submitted this supported their case of undue burden.
- Having now examined all the twists and turns in the attempts (by Mr Baldwin and Counsel for Biogen) to establish that the Skilled Team would have been able to implement the Patent, either at all or without undue burden, I can now state some conclusions:
i) On the basis of Construction 6, the Patent would have been invalid on classical insufficiency and uncertainty insufficiency, essentially for the reasons explained in Sandoz's submissions, as set out above.
ii) On the basis of Construction 5, the same conclusion applies i.e. the Patent is invalid because the aspects of the Gen2 assay which are not mentioned in the claim engage the same arguments that I have analysed above.
iii) On the basis of Construction 4, the same conclusion as for Construction 6 applies because of the issue that different populations have different medians - see [381]-[404] and [408]-[409] above.
iv) On these insufficiency issues, Constructions 1-3 all seem to come down to the same point - that, based on the level of JCV antibodies, there is some dividing line between low and high risk of developing PML. On this basis, the Patent would not be insufficient (but it would be obvious, for the reasons explained below).
- Sandoz's argument was that claim 1 encompasses the use of the claimed method in patients who have previously been administered immunosuppressants other than natalizumab. However, the claimed method is not suitable for use in this group of patients, because index values are not associated with risk of PML for such patients i.e. the hypothesis of the Patent, namely that "anti-JCV antibody positive patients [can] be further stratified for the risk of developing PML based on anti-JCV antibody titers" ([0216]), does not hold good for this patient population.
- Sandoz say that the problem is not simply that an index value other than 1.5 is appropriate to denote high risk of PML in the group of patients with prior immunosuppressant use. Nor can the risk for such patients be stratified by reference to an index value of 1.5, and then somehow modulated to take account of their prior immunosuppressant use. Instead, the problem is that anti-JCV antibody titers are not applicable at all when seeking to determine their level of PML risk.
- For the purposes of this argument, it does not matter how claim 1 is construed, because on any view it covers the application of a method of determining high risk on the basis of index values to patients with prior immunosuppressant use.
- In order to substantiate this point, Sandoz relied on three documents. First, they submitted this point was explained in a 2014 paper by Plavina et al., a paper by authors from Biogen (indeed Dr Plavina was one of the inventors) (emphasis added):
"Effect of Prior Immunosuppressant Use on Association between Index and PML Risk
Further analysis of the test data set identified a different relationship between index and PML for patients based on prior immunosuppressant use (Fig 3A). In patients with no prior immunosuppressant use, the index distribution was significantly higher for PML patients than for non-PML patients (median = 2.4 vs 1.4; p<0.0001). In contrast, index distribution was similar for non-PML and PML patients with prior immunosuppressant use (median = 1.6 for both groups; p = 0.82). A similar relationship was observed for the verification data set (see Fig 3B) and the combined data set (interaction p = 0.0158; Fig 4A). Thus, subsequent analyses of index and PML risk were limited to patients with no immunosuppressant use prior to natalizumab treatment. ..."
- Second, Sandoz say this was again confirmed by a 2017 paper by Ho et al. (again, Biogen authors) reporting an analysis of data from four clinical studies:
"Significant differences in index value between patients in the index cohort with and without PML were seen in patients without previous immunosuppressant use (p<0.0001) but not in those with previous immunosuppressant use (p=0.64, appendix)."
- That the method is not suitable for use in this patient group is made clear by the Physician Information and Management Guidelines that accompany Biogen's Tysabri product:
"In patients with prior IS [immunosuppressant], current data do not show an association between higher index and PML risk. The underlying biological explanation for this effect is unknown."
- When Dr Molyneux was asked about Plavina, he agreed that "titer is not helpful or useful, based on these data in a population of patients that have had immunosuppression, which is why the PIMG does not include titer levels in that population" and that "utility of the index values falls out for patients that have had prior immunosuppression". Dr Molyneux had referred to Plavina in his 2016 consensus paper (McGuigan [L/27]), making the point that the relationship between index value and PML risk was not seen in patients with prior immunosuppressant use, which is why he and his co-authors had excluded those patients from their guidelines. He also agreed that matters were confirmed by the Ho paper.
- Prof. Berger explained that antibody titers were not used in clinical practice to evaluate PML risk in patients who have received prior immunosuppressants since they are not useful for that purpose. He was not challenged on that evidence. This can be seen from the algorithm in the Biogen PIMG. Risk of PML for patients with prior immunosuppressant use depends not on index value but only on whether they are antibody positive and the duration of natalizumab treatment. As Dr Molyneux said, that was because it had since been "demonstrated that for patients with prior immunosuppressant use the antibody titer itself was not discriminatory and therefore the algorithm as listed does not give [a] break down according to the titer for patients with prior immunosuppressant use."
- I agree that this is a case in which there is a range relevant in the Regeneron sense (see [323] above) since the "variety does [...] significantly affect the value or utility of the claimed product or process in achieving its relevant purpose". At the priority date and today, prior immunosuppressant use was a known risk factor for PML. Dr Molyneux agreed that, in 2012, the Skilled Neurologist would have been aware that patients who had previously been treated with immunosuppressants were an important subgroup of the population of MS patients in terms of risk assessment.
- That is consistent with the fact that patients with prior immunosuppressant use comprise a significant proportion of MS patients. The Ho paper reports prior immunosuppressant use in patients from the STRATIFY-2, STRATA, TOP and TYGRIS studies (in total over 37,000 patients). On average, 14% of patients from that cohort had previously been administered immunosuppressants, with an incidence of 20% in the TYGRIS study. Dr Molyneux agreed that patients with prior immunosuppressant use were a significant subset of the cohort of patients reported in the Ho paper, though he made the point that immunosuppressant use was lower in the UK than in the US and continental European countries.
- Biogen advanced three points in response but, for the reasons explained below, they have no merit and do not dent the facts established by Sandoz set out above.
- First, Biogen's written evidence was aimed at establishing that Example 6 and Figure 13 made it plausible at the priority date that the method of claim 1 could be used for patients with prior immunosuppressant use as well as those without. However, as Sandoz submitted, that addresses the wrong question: the complaint is that there exists a significant group of patients for whom in fact the method does not work.
- Second, Biogen submitted in §§134-136 of its opening skeleton that the attack "rests on the implicit premise that the claim is to a complete method of evaluating the risk of PML" whereas the claim is "to a method of assessing risk of developing PML on the basis of a single factor, i.e. anti-JCV antibody titre, not to a method of assessing the patient's overall risk." However, I agree there is no such premise in Sandoz's case. The claim requires a patient to be determined to be at high risk of PML if their index value is > 1.5. But for patients with prior immunosuppressant use, there is no link between risk of PML and index value. A clinician cannot, and does not, determine risk of PML for such patients based on index values.
- Third, Biogen developed a case that the cohort of patients who have been administered immunosuppressants is a small one. Dr Molyneux described the number of patients with prior immunosuppressant use in the UK as being "tiny". In §203 of his first report he had described it as a "relatively small proportion". Sandoz drew attention to the fact that Biogen declined to adduce any evidence as to the actual proportion of such patients in the UK, either as at 2012 or at any other time.
- However, I agree that a purely quantitative approach is wrong. It was common ground that the prior immunosuppressant use group is an important one from a risk assessment perspective. That view recognises that there were a sufficient number of patients in this cohort for clinicians to be aware of their specific treatment needs. They clearly did not fall below the radar, and so cannot be dismissed as de minimis. Indeed, they are expressly addressed in the current risk assessment algorithm (as they were in the algorithm current in 2012).
- Sandoz also submitted it was wrong to focus on the UK numbers. The Patent is based upon data from patients participating in a clinical trial in the US (Stratify-1), which included patients with prior immunosuppressant use. Example 6, with reference to Figure 13, presents its results as being applicable to patients with prior immunosuppressant use regardless of the numbers involved (in fact Stratify-1 only included 3.8% of such patients - see Table 2). As they submitted, it would be contradictory to support an invention by reference to such US data, including on low numbers of IS+ patients, and then selectively take a UK-specific approach in order to avoid the consequences of the fact that the conclusions drawn from that data set have turned out to be wrong.
- Accordingly, I find the patent invalid on breadth of claim insufficiency, and this applies irrespective of the correct construction.
- Sandoz contended that the subject-matter of the claims (alternatively any non-obvious subject matter) is non-technical in nature and does not amount to an invention within the meaning of the Act, whether because it consists of a scheme, rule or method for performing a mental act, or the presentation of information, or otherwise.
- The arguments were addressed by both sides by reference to the four-stage test in Aerotel:
i) properly construe the claim;
ii) identify the actual contribution;
iii) ask whether it falls solely within the excluded subject matter; and
iv) check whether the actual or alleged contribution is actually technical in nature.
- Sandoz addressed the issue on the basis of Construction 6, but the difference with Construction 5 makes no difference. For their part, Biogen addressed the issue on the basis of Construction 3, as far as I could tell i.e. the claim is not limited to any specific antibody titre, but the 1.5 value in the claim is a means of expressing a dividing line between sero-positive patients at higher and lower risk of developing PML.
- The parties were agreed that a series of features in the claim were all disclosed or obvious over the prior art and so were not the technical contribution of the claim: essentially the use of an ELISA employing an HPVLP capture antigen, controls and a calibrator to determine an anti-JCV antibody titre expressed as an index value.
- In their opening skeleton, Sandoz identified the nub of the dispute as follows:
"The actual contribution lies in determining a patient to be at high risk (whatever that is) of developing PML at an index value of> 1.5 (where that index value corresponds to a certain anti-JCV antibody titer, whatever that is). That is not technical in nature. It involves taking information presented by the assay and performing a mental act using that information, namely assessing risk of PML as being high. It follows that the subject-matter of the claims falls foul of the s.1(2) exclusion."
- In their response, Biogen chose to adopt (for the purposes of argument) what they referred to as 'Sandoz's miserly construction of the claim' which I understood to be a reference to Construction 6. Even on that basis, Biogen submitted the contribution is the specific antibody titre associated with the preliminary clinical cut point presented in Example 6. Biogen submitted that contribution may be narrow, but it is plainly technical in nature. I agree. Professor Berger freely acknowledged that it represents a useful solution to the problem of stratifying the risk of PML within the antibody positive population. He went so far in cross-examination as to describe it as a '...very, very impressive and strong result...'.
- Accordingly, on either of Constructions 5 or 6, the excluded subject matter allegation fails.
- On their construction (i.e. Construction 3), Biogen argued that the technical contribution is similar in kind but broader in scope. Biogen said it resides in the breakthrough idea of using antibody titre to stratify the risk of PML within the antibody positive population at all, contending that contribution is plainly technical in nature for the same reasons.
- On this point, Sandoz posed the rhetorical question: if the 1.5 index value in the claim is just an arbitrary number, then what is the technical contribution to the art in the claim? To put it another way, what in the claim adds to human knowledge compared to Gorelik / WO369?
- If any of Constructions 1-3 was to represent the correct construction of Claim 1, then I find that the Patent makes no technical contribution to the art over WO369 which, as explained above, disclosed the broad concept of a division between high and low risk of developing PML (i.e. high/moderate in WO369), based on the level of JCV antibodies.
- Sandoz contends that the Patent is invalid for added matter for two reasons, namely those pleaded in §7(c) and in §7(d)-(g) of the Amended Grounds of Invalidity. The relevant comparator is the parent application for the Patent ("the PCT").
- The §7(c) point relates to claim 8, which reads as follows:
"The method according to any one of claims 1 to 7, wherein the patient determined to be at high risk of developing PML is determined to be at higher risk of developing PML if the patient has received natalizumab for longer than 24 months and has not previously received a non-anti-VLA-4 immunosuppressant therapy, wherein the non-anti-VLA-4 immunosuppressant therapy is selected from mitoxantrone, methotrexate, azathioprine, cyclophosphamide, mycophenolate, anti-CD20 therapy, anti-CD11a therapy, and mycophenolate mofetil."
- Sandoz say the PCT does not disclose such a method.
- To aid analysis Sandoz adopted the following nomenclature: relative risk of PML for a given patient group "R", anti-JCV antibody positive "Ab+", anti-JCV antibody titer or percent inhibition "J", the pre-determined level "X", specified prior immunosuppressant use "IS+" (and its absence "IS-") and extended treatment with anti-VLA-4 therapy "V+" (and its absence "V-").
- In the Defence, [5E] Biogen relied upon the passage at p.15 l.27 - p.16 l.12 of the PCT. Sandoz made submissions in their opening skeleton explaining why such reliance was misplaced. Biogen did not rely upon this passage in their opening skeleton. In their closing, Biogen said they had nothing to add to their opening skeleton, save to make clear that [5E] of their Defence was maintained, so I will briefly address this passage (in which I have labelled particular phrases).
"In one embodiment, a method of evaluating a patient as described herein, such as to determine an anti-JCV antibody titer or percent inhibition, can further include assessing other measures of risk predictors. For example, a method of evaluating a patient can further include: (a) determining if the patient has received extended treatment with an anti-VLA-4 therapy (e.g., longer than 24 months); or (b) determining if the patient has received a specified non-anti-VLA-4 immunosuppressant therapy (e.g., mitoxantrone or other therapies in the last 2, 3, 5 years or ever in the patient's life). [1. The relative risk of PML for a patient who has an anti-JCV antibody titer or percent inhibition above a pre-determined level but has no specified prior immunosuppressant use and has not had an extended treatment with an anti-VLA-4 therapy] is less than [2. the relative risk of a patient who has an anti-JCV antibody titer or percent inhibition below a pre-determined level and has specified prior immunosuppressant use or an extended treatment with an anti-VLA-4 therapy], which is less than [3. the relative risk of a patient who has an anti-JCV antibody titer or percent inhibition above a pre-determined level and has specified prior immunosuppressant use and an extended treatment with an anti-VLA-4 therapy]."
- As Sandoz submitted, this passage says: 1. R(J>X, IS-, V-) < 2. R(J<X, IS+ / V+) < 3. R(J>X, IS+, V+) whereas claim 8 relates to the group (J>X, IS-, V+), which does not correspond to any of the groups considered in the passage relied on by Biogen. Further, the passage relied on by Biogen does not disclose that the pre-determined level is an index value of 1.5, or that the anti-VLA-4 therapy is natalizumab, or that the non-anti-VLA-4 immunosuppressants are those specified in claim 8. In fact, the PCT does not contain, at any point, the list of non-anti-VLA-4 immunosuppressants specified in claim 8. So, as Sandoz submitted, it is not a great surprise that Biogen did not rely upon this in their Opening.
- Instead, Biogen relied (see [210] of its opening skeleton) on the paragraph at p.20 l.23 - p.21 l.2 of the PCT, and in particular the underlined parts:
"In an embodiment, a patient who has received an anti-VLA-4 therapy, such as natalizumab, for longer than 24 months (e.g., for 25 to 48 months, such as 26, 30, 36, 42 25 or 48 months or longer), and who has not received prior treatment with an immunosuppressant therapy other than an anti-VLA-4 therapy can be determined to be at a higher risk for PML. A patient at a higher risk for PML can have a risk of about 0.37/1000 or greater, e.g., about 4.3/1000 patients. The patient can accordingly be determined not to be a candidate to receive further treatment with an anti-VLA-4 therapy, or can be determined to be a candidate to receive treatment with an anti-VLA-4 therapy accompanied by more frequent monitoring."
- Biogen say that the allegation that this embodiment does not disclose (i) that the patient has been determined to be at high risk of PML by reason of having an anti-JCV antibody index value of > 1.5, and (ii) has not received prior treatment with the immunosuppressants listed in claim 8, depends on taking the embodiment out of its proper context.
- In this regard, Biogen say this passage forms part of the 'Summary of the Invention' beginning at p.1. The broad scheme of this summary they say is to provide:
i) general teaching about the assay used determine anti-JCV antibody titres (p.1, line 26 - p.10, line 22);
ii) general teaching about how the results of the assay may relate to the estimated risk of PML (p.10, line 23 - p.17, line 18);
iii) general teaching about combining the results of the assay with other risk factors, including length of treatment with an anti-VLA-4 therapy (e.g. natalizumab) and prior treatment with a non-anti-VLA-4 immunosuppressant (p.10, line 19 - p.29, line 26).
- Biogen say the disclosures of these three sub-sections build upon one another: sub-section iii) is able to discuss how anti-JCV antibody titre can be combined with other risk factors because sub-section ii) has previously disclosed anti-JCV antibody titre as a risk factor in its own right and sub-section i) an assay for detecting anti-JCV antibody titre. Accordingly, Biogen submit that these general teachings are therefore capable of being combined without adding matter.
- Biogen say that it follows that the p.20, line 23 embodiment (which forms part of sub-section iii) can and should be read in combination with the methods of relating the results of the assay to the estimated risk of PML disclosed in sub-section ii). These include the method of the patient being '... determined to be at higher risk of PML if (i) the anti-JCV antibody ...index value... is determined to be > 1.5...' (p. 17, lines 8 - 18). The combination of the p.20, line 23 embodiment with the patient having been determined to be at high risk of PML by reason of having an anti-JCV antibody index value of > 1.5 is not therefore new information.
- As for Sandoz's objection (ii), Biogen rely on a later passage of the PCT which lists the non-anti-VLA-4 immunosuppressants listed in claim 8 (p.35, lines 4 - 9). Although nominally a part of the 'Detailed Description' beginning at p.31, the Skilled Team would plainly understand this passage in context to exemplify the non-anti-VLA-4 immunosuppressants discussed earlier in the document. Accordingly, the combination of the p.20, line 23 embodiment with this list is not new information.
- The 'Summary of the Invention' section in the PCT extends from p1-30. In a familiar manner, it sets out a whole series of embodiments and aspects, each of which sets out one or more specific features. It is apparent that where the patentee wanted to disclose a particular combination, it did so.
- As Sandoz pointed out, this passage (i.e. as quoted at [457] above) does not relate to patients who are determined to be at high risk of PML by reason of having an anti-JCV antibody index value of > 1.5. This passage appears in a section of the PCT at p.18 l.12 - p.22 l.9 which discusses risks based on (i) whether the patient has received natalizumab treatment for longer than 24 months or not, (ii) whether the patient has received non-anti-VLA-4 immunosuppressant therapy or not and (iii) whether the patient has tested positive or negative for JCV.
- Thus at p.18 ll.12-19, the PCT states that patients with the lowest risk are typically those who have received anti-VLA-4 therapy for 24 months or less ("V-"), who have not previously received immunosuppressant therapy ("IS-") and test negative for anti-JCV antibodies ("Ab-"); and those with highest risk are those who had received anti-VLA-4 therapy for more than 24 months ("V+"), have previously received immunosuppressant therapy ("IS+") and are anti-JCV antibody positive ("Ab+"). Then, from p.18 l.25 - p.22 l.30, the PCT then considers patient groups answering different permutations of those parameters.
- The permutation identified at p.20 l.23 - p.21 l.2 (i.e., the V+ / IS- / Ab+ group) are said to be at 'higher risk' of PML - specifically, a risk of 4/1000 patients.
- In cross-examination, Dr Molyneux agreed (by reference to [0074] of the Patent (which is similar but does include Ab+)) that this passage (like the surrounding paragraphs) merely documented the common general knowledge (note that the risk level is essentially that stated for this group in the PIMG current in 2012, which provided a risk of ~3.9/1000 for this patient group - see Figure PM10 in Molyneux 2). Sandoz also point out that this is not the only part of the Patent that just reports matters that were common general knowledge - Example 1 culminates in the algorithm in Figure 3 which had already appeared in Kappos.
- Accordingly, Sandoz submitted that all p.20 l.23 - p.21 l.2 is doing is stating that which was part of the common general knowledge regarding risk assessment dependent on whether patients were antibody positive or negative. In context, I agree that Biogen are wrong to say that it can somehow be combined with other teachings in the document and in particular teachings about determination of risk dependent on whether a patient has an index value of > 1.5.
- The problem for Biogen is that they have been unable to point to any passage in the PCT in which risk assessment based on an index value >1.5 has been considered for patients who (a) have had natalizumab treatment for more than 24 months and (b) have not had prior non-anti-VLA4 immunosuppressant therapy.
- Biogen also rely on p.17 ll.8-18 (see §214 of its opening skeleton) but they do so selectively. In that passage, the patient is determined to have a higher risk of PML if:
"(i) the anti-JCV antibody titer as indicated by index value or nOD is determined to be > 1.5 and the percent inhibition value is determined to be > 70%, or (ii) the patient showed an increase in index, nOD or titer by 2-fold from a previous test."
- First, there are two options as to how higher risk is to be determined. Biogen do not explain why they alight on option (i) rather than option (ii). Second, even taking option (i), the disclosure requires both an nOD over 1.5 and a percent inhibition value over 70%.
- The second point relating to this added matter attack is that p.20 l.23 - p.21 l.2 does not identify patients who have not received prior therapy with the particular non-anti-VLA-4 immunosuppressant therapies which are specified in claim 8.
- For this, Biogen rely on a passage which is found at p.35 ll.4-9 of the PCT which states:
"Prior immunosuppressant therapies, other than anti-VLA-4 therapy, that will be indicative of an increased risk of PML can include prior treatment with antineoplastics, immunosuppressants or immunomodulators, such as one or more beta-interferon or glatiramer acetate. Exemplary immunosuppressants include, e.g., mitoxantrone, methotrexate, azathioprine, cyclophosphamide, and mycophenolate, anti-CD20 therapy (e.g., rituximab), an anti-CD11a therapy (e.g., efalizumab), or mycophenolate mofetil."
- However, the passage at p.20 l.23 - p.21 l.2 states that a patient is at higher risk where inter alia that patient has not received non-anti-VLA-4 immunosuppressant treatment. In other words, that associates risk with having received any such immunosuppressant. By contrast, claim 8 identifies higher risk for patients who have not been administered with certain specific immunosuppressant treatments. The claim teaches for the first time that risk is associated with certain treatments (i.e. those specified) rather than the fact of having taken a non-anti-VLA-4 immunosuppressant before. Those are conceptually different things.
- To look at the point another way, claim 8 includes patients who have been previously administered with forms of non-anti-VLA-4 immunosuppressant treatment other than those specified, whereas p.20 l.23 - p.21 l.2 does not. Claim 8 thereby discloses for the first time a method for determining patients to be at high risk if they (i) have an index value > 1.5, (ii) have been treated for natalizumab for more than 24 months and (iii) have not been treated with a form of non-anti-VLA-4 immunosuppressant treatment other than those specified.
- For all these reasons, I agree that claim 8 adds matter over the disclosure of the PCT.
- Sandoz explained this point in the following way: in the PCT an association between an index value of >1.5 and increased risk of PML is disclosed by reference to Example 6, in which the data is generated using the Gen2 assay. However, the Patent introduces claim 1 (and the corresponding [0005]) and so discloses the use of an assay which does not have all the features of the Gen2 assay to determine that a patient is at high risk of PML if their index value is >1.5 (so long as, in that assay, an index value of 1.5 corresponds to the same anti-JCV antibody titer as in the Gen2 assay). Therefore the Patent teaches that a patient can be determined to be at high risk of PML if they have an index value of >1.5 even if an assay which does not have all the features of the Gen2 assay is used. This is, it is submitted, an intermediate generalisation.
- Biogen submitted that this attack was misconceived. Harking back to their argument on insufficiency they submit that the Skilled IDS would not understand all the features of the Gen2 assay to be a necessary part of the invention. Biogen argue that, armed with the teaching of Figs 12 & 13, the Skilled IDS has the option of working the invention by setting up a different assay that produces a comparable result. This, it seems to me is a reprise of part of the insufficiency arguments. However, whether this is an intermediate generalisation depends on the correct construction. On Construction 6, it would not be (i.e. the Skilled IDS would understand that the invention is wholly dependent on the Gen2 assay), but on Construction 5, it is.
- Biogen detected, correctly, there were three routes to obviousness apparent in Sandoz's evidence. I have dealt with the first above - where I determined that Prof Berger's impression argument was not a starting point for an obviousness argument. The second was a virological argument, said to be set out in Dr Dugan's evidence. Since her evidence was withdrawn, I have not paid any attention to it, except for the passages which were agreed to by Prof Roy. I gather it was another possible starting point providing some sort of hypothesis linking anti-JCV antibody levels to the risk of PML. It is unnecessary to set out all the cross-examination relating to this. The end point was summarised by Biogen as follows:
'This oral evidence from the neurologists (coupled with the withdrawal of Dr Dugan's report) establishes that there existed no theoretical reason in the CGK relating to the virology or immunological response to JCV, or the pathogenesis of PML, for the Skilled Team to suspect a link between anti-JCV antibody levels and the risk of PML, and no motivation to investigate such a link. To the contrary, PML was known to be associated with AIDS and the use of immunosuppressive drugs which are consistent with poor immune responses which might be expected to result in lower antibody titres.
Accordingly, no such pre-existing suspicion or motivation can be ascribed to the Skilled Team when they come to the prior art.'
- I proceed on that basis. That leaves Sandoz's third route, which I address below.
- The parties made brief submissions on the legal principles. Naturally I was reminded of the review by the Supreme Court of the basic legal principles in Actavis v ICOS [2019] UKSC 15 at [52]-[73]. Biogen emphasised [65] in particular:
"First, it is relevant to consider whether at the priority date something was "obvious to try", in other words whether it was obvious to undertake a specific piece of research which had a reasonable or fair prospect of success: Conor v Angiotech (above) para 42 per Lord Hoffmann; MedImmune Ltd v Novartis Pharmaceuticals UK Ltd [2012] EWCA Civ 1234; [2013] RPC 27, paras 90 and 91 per Kitchin LJ. In many cases the consideration that there is a likelihood of success which is sufficient to warrant an actual trial is an important pointer to obviousness. But as Kitchin LJ said in Novartis AG v Generics (UK) Ltd [2012] EWCA Civ 1623, para 55, there is no requirement that it is manifest that a test ought to work; that would impose a straightjacket which would preclude a finding of obviousness in a case where the results of an entirely routine test are unpredictable. As Birss J observed in this case (para 276), some experiments which are undertaken without any particular expectation as to result are obvious. The relevance of the "obvious to try" consideration and its weight when balanced against other relevant considerations depend on the particular facts of the case."
- For their part, Sandoz drew attention to these points:
i) First, it is well established that there can be no invention in doing what is suggested in the prior art (Lilly v Pfizer [2002] EWCA Civ 1 at [53]; see also Lewison LJ in Medimmune v Novartis [2012] EWCA Civ 1234 at [184]-[185]) unless there is an established technical prejudice against that idea - the so-called "lion in the path" (Pozzoli v BDMO [2007] EWCA Civ 588 at [24]-[29]). Sandoz did not understand Biogen to contend that there is any such technical prejudice in this case.
ii) Second, if a claim feature is arbitrary, then it cannot assist in defending an obviousness case (see Optis v Apple [2020] EWHC 2746 at [207]). More generally, an inventive step requires a technical contribution to the art (see e.g. Takeda UK v Hoffmann-La Roche [2019] EWHC 1911 at [203]-[207].)
- In closing, Sandoz submitted that their case of obviousness was compelling, whether based on Gorelik or WO369, but that the case over WO369 was unanswerable because WO369 identifies the very same hypothesis as that set out in the Patent and expressly discloses stratifying patients into sub-groups with differing PML risk levels based on their antibody levels.
- Sandoz summarised their case as follows: there was clear motivation at the priority date to explore further the risk factors surrounding PML, in particular to focus on whether further risk stratification of anti-JCV antibody positive patients was possible. It is common ground that the Skilled Neurologist would have been strongly motivated to investigate the clinical utility of the two-step assay disclosed in Gorelik and WO369 on a larger cohort of patients, including PML patients. As part of that study, nOD and % inhibition values would be collected and the correlation between antibody levels and PML risk would emerge from those data. Identification of the threshold for high-risk patients would be a matter of clinical judgment based on the Skilled Neurologist's attitude to acceptable risk and the data obtained from the patient population studied.
- Throughout, Sandoz submitted that it is important to remember that it is common ground that the notional Skilled Team comprises a Skilled Neurologist, a Skilled IDS and a Skilled Virologist. Further, it is also common ground that the Skilled IDS could produce an immunoassay similar to that described in Gorelik and WO369 to test the level of antibodies in a patient sample. Sandoz submitted that the obviousness case turns on the question of whether it would be obvious to the Skilled Neurologist to use such an assay to investigate antibody levels and PML risk.
- Thus, an important part of Sandoz's case depended on the notional nature of the Skilled Team. Whereas in the real world, neurologists were only receiving results from the testing lab in the form of a positive or negative result, the notional Skilled Team would have their own assay which would be generating nOD and % inhibition results. This leads to the issue which I have to consider further below - where would the Skilled Team obtain samples or data.
- Sandoz chose to emphasise in their closing some CGK points which they said were of particular importance to the obviousness analysis. I set them out here.
- First, natalizumab was a "game changing development in the treatment of MS" that acted like a switch in effectively turning off patients' relapses. As Dr Molyneux vividly explained:
"Having for a decade watched patients that I had been managing have relapses, the distress that is caused to them by that, the impact on their family life, the loss of income and slowly accumulate disability, it is incredibly helpful to have a highly efficacious drug that can meaningfully reduce the relapse rate in some pose Phase IV studies by up to 98%. So it effectively, for the majority of patients, switched their MS off. It gave patients their lives back. It had an incredibly profound effect on the cohort of patients I was privileged enough to treat."
- Both clinicians and their patients were therefore keen to use natalizumab if possible. However, it was recognised that treatment with natalizumab came with a small, but nevertheless very serious risk, of developing PML. The informed assessment of PML risk was therefore extremely important for MS patients.
- It was sensible and obvious for the Skilled Neurologist to seek to improve the understanding of PML risk and the factors that were associated with PML risk, and the Skilled Neurologist would want to know as much as possible about what the risk factors for PML were.
- By the priority date, three independent risk factors were known: prior infection with JCV, duration of natalizumab treatment and prior treatment with immunosuppressants.
- Prior infection with JCV was a necessary condition for the development of PML and it was known that the presence of antibodies to JCV in a patient's serum indicated a prior JCV infection. By the priority date, Biogen had made the STRATIFY JCV testing service available. Clinicians knew that the STRATIFY JCV test was a two-step assay in which the first step was an ELISA to detect antibody levels in a sample and the second step was a confirmatory test of inhibition in those samples with indeterminate antibody levels. However, clinicians were only told whether their patients tested positive or negative for anti-JCV antibodies.
- There was a recognition that it was important to obtain as much information as possible about MS patients receiving treatment with natalizumab. Recording of patients' anti-JCV antibody test results together with information about other risk factors (i.e. time that natalizumab treatment started and prior immunosuppression) as well as whether the patient went on to develop PML was routine. Dr Molyneux's evidence was that it would "seem extraordinary" not to keep such records and there were a number of national and international registries established to identify any patterns in Phase IV studies.
- Seropositivity rates were known to be high at the priority date, with the literature reporting rates in the range 50 to 80%. Accordingly, if no anti-JCV antibody positive MS patients were treated with natalizumab, a large number would miss out on this ground-breaking treatment. There was therefore a real incentive amongst Skilled Neurologists to find a way to improve the assessment of an individual's risk of PML at the priority date.
- Despite the risk of PML, some seropositive patients did decide to commence or continue natalizumab treatment. Conversely, others would decline treatment if they received a positive antibody test result. Accordingly, there was a desire to have a means to better assess a patient's personal risk of PML at the priority date and to differentiate the levels of risk within the anti-JCV antibody positive population. This was described by Prof. Berger at §65 of his first report and agreed with by Dr Molyneux, subject only to the caveat that it was not known at the priority date how to better assess a patient's risk:
"Q. Okay. Do you agree with what Professor Berger says there?
A. So, I would absolutely agree with what he says around the fact that a significant proportion of patients who tested JCV positive would take a decision around their own risk of PML, and either the neurologist, the very risk-averse neurologist might persuade the patient to come off treatment or the patient would take their own view. There is always a debate in terms of shared decision-making about how much the views of the neurologist would play into the decision, how that decision is played out. Broadly, I would accept that a neurologist would regard it as helpful to clarify the risk as much as was possible given the evidence of that was available.
Q. Right. So you would agree with him that there was a desire to have a means to better assess a patient's personal risk of PML and differentiate the levels of risk within the antibody positive population so that a more informed decision could be made about the risks and benefits of treatment?
A. I agree, but I would caveat that by saying I am not sure the neurologist at that point would have known how to better assess the patient's personal risk at that point."
- Sandoz's third route comprised the following points.
- It was common ground that the Skilled Neurologist reading Gorelik at the priority date would have understood it to be reporting the research that led to the STRATIFY JCV assay that was made available about a year before the priority date.
- It was also common ground between the assay experts that the Skilled IDS could produce an assay of the same general format as described in Gorelik, namely one that measures anti-JCV antibody levels based on nOD levels in an indirect ELISA and determines a % inhibition figure in a competitive ELISA. It is also not suggested by Biogen that there is anything inventive in the particular nOD values specified in claim 1 for the positive and negative controls: a point confirmed by Mr Baldwin in cross-examination.
- Accordingly, so Sandoz submitted, the only issue on obviousness is whether it would have been obvious for the Skilled Team to use a Gorelik-type assay so as to reveal the association between antibody levels and risk of PML. Sandoz submitted it was, on the basis that the evidence showed that the Skilled Neurologist would have been motivated to use a Gorelik-type assay on a large number of patients to test its clinical utility as a tool for stratifying MS patients for risk of PML. As part of such testing, the Skilled Team would collect data, including nOD values and % inhibition values, from MS patients including those who had gone on to develop PML. The association between anti-JCV antibody levels and the risk of PML would emerge as those data were collected.
- In the context of Gorelik, there did not seem to be a dispute that there was motivation to investigate further the risk of developing PML. It was CGK that there was a desire to have a means to better assess a patient's personal risk of PML at the priority date as opposed to simply applying the three known independent risk factors of prior JCV infection, duration of natalizumab treatment and prior immunosuppressant treatment. The Skilled Neurologist was seeking a way of further differentiating risk within the seropositive population. As Prof. Berger explained:
"...one of the limitations of the anti-JCV antibody testing provided by Biogen in April 2012 was that it could only give a positive or negative result. Although it was useful to identify anti-JCV antibody positive patients, it did not enable further differentiation between those patients in terms of their risk of PML. The Skilled Neurologist would be aware that a large proportion of the population test positive for JCV infection (over 50%). Given the high past infection rate, the rarity of PML and the limited number of other treatment options available, there was a desire among patients and clinicians to continue to use natalizumab in anti-JCV antibody positive patients. This was driven in particular by the severity of many patients' MS symptoms, which were often life limiting and could be improved by natalizumab treatment. At the same time, treatment needed to be undertaken in the safest manner possible and therefore there was a motivation to further understand and stratify the risk of PML in JCV positive patients treated with natalizumab."
- Accordingly, even if it was not known what risk factors might emerge, there was a desire to continue to investigate risk so as to be able to better counsel patients. Both clinical experts agreed that the Skilled Neurologist would be interested in Gorelik and would want to take it forward for this very reason. Dr Molyneux's evidence was that there would have been interest in research that "led to a better understanding of our ability to stratify patients in terms of their risk of PML".
What would be done?
- It was common ground that the Skilled Neurologist would use a Gorelik-type assay to test a larger number of patients, including PML patients:
"The Skilled Neurologist would have wanted to review further data from a larger number of patients (both PML and non-PML) than were evaluated in Gorelik, which would have been collected as a matter of good clinical practice in any case...This would essentially be a continuation of the studies described in Gorelik" Berger 1 §80
"...after reading Gorelik the Skilled Neurologist would be interested in further assessing the value of the assay in predicting PML risk in the clinical setting. They would have been motivated to test the binary assay that is described in the paper on a larger number of MS patients and, particularly, in more samples taken from patients who would go on to develop PML, ideally including prospective study data..." Molyneux 1 §186
- In his evidence quoted above, Dr Molyneux referred to Gorelik being a "binary assay" meaning that it produces a positive or negative result. This was a point that he made several times in cross-examination [e.g. T2 120/10-25 & 122/3-20]. Biogen's STRATIFY JCV assay service only provided clinicians with positive or negative results. However, the Skilled Neurologist is part of a team that includes the Skilled IDS. The Skilled Team is therefore able to produce its own assay and there is, therefore, no question that the Skilled Team will have access to the underlying nOD values [T2 126/9-24] (see also [T2 120/17 - 121/2-5] where Dr Molyneux accepted that Gorelik discloses levels of antibodies, measured as an nOD value, and provides the specificity of antibodies, measured as % inhibition, and shows that there is a spread of both nOD values and % inhibition values in the samples tested).
- Dr Molyneux confirmed that the Skilled Neurologist would want to test "thousands" of patients, including patients who went on to develop PML. He said that "ideally" this would include prospective study data, but agreed that retrospective samples could also be used.
- Following cross-examination, Sandoz submitted that it appeared that the dispute between the parties came down to the question of whether the Skilled Neurologist would examine the nOD and % inhibition levels produced by the assay or whether, as Dr Molyneux suggested, the Skilled Neurologist would be content simply to use that same information to categorise patients as positive or negative and pay it no further attention.
- Sandoz's position was that the Skilled Team would collect nOD and % inhibition levels, together with details of which patients had developed PML, and would examine that data, e.g. in the way shown in Figs. 3 and 6 of Gorelik and Figs. 11 and 12 of the Patent.
- Dr Molyneux's insistence that the Skilled Neurologist would not be interested in the data underlying the positive / negative result was not credible, and the point was not put to Prof. Berger. The Skilled Neurologist was seeking a way of further differentiating risk within the seropositive population. A Gorelik-type assay would produce data which discriminates between the seropositive population in terms of nOD and % inhibition. Why would the Skilled Neurologist not look at the data to see whether there is an association between those factors and the risk of PML? Sandoz suggested that it may have been the case that Dr Molyneux's view was conditioned by the fact that he was a prescribing neurologist who did not conduct clinical research, and had seen the Skilled Neurologist as such a person.
- In any event, Sandoz submitted that Gorelik provided further reasons to be looking at nOD values. One of the things that the Gorelik authors identify as requiring further study is the false negative rate, which requires consideration of nOD values. Even if, as Dr Molyneux suggested during cross-examination, optimisation of the assay to reduce the false negative rate is more for the Skilled IDS, the Skilled Neurologist works as part of a team that includes the Skilled IDS and the clinical utility of the assay will depend in part on its false negative rate.
- Another aspect identified by Gorelik is that of the longitudinal stability of the results generated; they note that some patients seroconvert (i.e. become seropositive) whereas others serorevert (i.e. become seronegative). Prof. Berger explained (without challenge) that the Skilled Neurologist:
"..would also want to conduct longitudinal testing of seropositive patients to see if any pattern could be seen in their antibody levels over time, particularly in patients who went on to develop PML as this might also provide a way to stratify patient risk within the anti-JCV antibody positive group."
- Dr Molyneux agreed that one interpretation of the seroconversion/reversion results in Gorelik was variation around a cut-point, although other interpretations might be possible. One way to investigate this would be to collect nOD values and % inhibition values as part of the study that the Skilled Neurologist was motivated to carry out.
- If the Skilled Team proceeded as Sandoz contends, the association between higher antibody levels, expressed as nOD, and PML risk would emerge from such data.
- It would then simply be a matter of clinical judgment as to where to put the cut-point above which patients would be identified as being at high risk of PML, balancing the desire to treat as many patients as possible against the risk of falsely identifying patients who will go on to develop PML as being at low risk. As Prof. Berger explained in his written evidence, precisely where the line for would be drawn would depend on the Skilled Neurologist's appetite for risk and the data obtained from the patient population. As such, drawing the line so that the threshold sits at the same antibody level as that identified in Example 6 of the Patent using the Gen2 assay is just one of several obvious thresholds to choose.
- Sandoz also rely on secondary evidence arising from two papers - Trampe and Major.
- Despite Dr Molyneux's evidence that it would not be obvious to look at the nOD and % inhibition data when testing a Gorelik-type assay on a larger cohort of patients, Sandoz submitted that was, in fact, precisely what a group of neurologists did do before the priority date as evidenced by the Trampe paper, which was submitted in October 2011.
- Trampe summarised their objectives as follows:
"To better understand whether epidemiologic factors, including geography, gender, age, immunosuppressive pretreatment, and duration of natalizumab treatment influence the seroprevalence and levels of anti-JCV antibodies and the utility of serostatus for PML risk stratification, a large cohort of German patients with MS treated with natalizumab was investigated for anti-JCV antibodies. In addition, samples available from 10 natalizumab-treated patients collected before or at PML diagnosis were analyzed in a blinded fashion."
- Dr Molyneux agreed that this was the kind of study which the Skilled Neurologist would want to have done, save that he said they would not have been interested in antibody levels.
- The authors conducted a longitudinal analysis of serostatus and antibody levels to address the question of whether seroconversion and seroreversion were due to fluctuations of levels around the cut-points. They also examined the antibody status and levels of the PML patients and compared them to those of non-PML patients. The association between antibody levels and PML risk emerged from the data.
- Dr Molyneux's point was that the study concept and design for the Trampe study involved Dr Goelz, who had worked with Biogen and was a co-author on Gorelik. Sandoz submitted that this did not undermine their point because this was someone who was familiar with Gorelik who has decided to do precisely the kind of study Sandoz contended would have been obvious to do given Gorelik. As Sandoz pointed out, Dr Goelz is not an inventor on the Patent. Further, none of the other authors of the Trampe paper is a named inventor, and none of the named inventors was an author on the Trampe paper. Accordingly, Sandoz submitted there is nothing to suggest that Dr Goelz's study concept and design was influenced by any of the inventors of the Patent.
- However, as Biogen suggested, it is likely that the study reported in Trampe was able to be conducted because of the link to and the cooperation of Biogen.
- Sandoz suggested that Dr Molyneux's view may have been based on the fact that he personally had not heard anybody, around the priority date, propose that there might be a relationship between specific antibody levels and PML risk. Sandoz had two answers to that suggestion:
i) First, as explained above, it was not necessary to Sandoz's obviousness case for the Skilled Neurologist to have considered that there may be an association between antibody levels and PML risk: there were other reasons to look at nOD levels (e.g. to investigate the degree of seroconversion and seroreversion, as identified in Gorelik and investigated further in Trampe). Sandoz's point was that it is unrealistic to suggest that the Skilled Team carrying out a study using a Gorelik-type assay would not look at antibody levels measured (expressed as nOD) and interrogate that data for information about PML risk.
ii) Second, Sandoz submitted that Dr Molyneux's personal experience does not necessarily reflect that of the Skilled Neurologist. By the priority date he had been working for many years as a clinical neurologist rather than a neurologist involved in clinical research and Sandoz suggested it may be the case that he simply did not come across such discussions. Certainly, such discussions were taking place: the Major paper records a roundtable at the Cleveland Clinic in January 2011, in which rising anti-JCV antibody levels were identified as a risk factor for PML, as Dr Molyneux accepted [T2 161/10 - 164/12].
- Before proceeding further, I will set out Sandoz's case on WO369, not least because I understood Biogen's response to apply to both cases equally.
- I have resolved the dispute over what WO369 disclosed to the Skilled Team, in particular at p.6 ll.15-27, at [179] above. As Prof Berger said, the Skilled Team would understand that patients in the high and moderate risk categories would be positive for anti-JCV antibodies, and the division between these categories was based on the level of anti-JCV antibodies. However, the dismissive attitude of Biogen and Dr Molyneux also raises an issue over motivation to proceed based on this disclosure.
- Biogen's position was that Dr Molyneux's evidence in chief on this point was clear and unshaken in cross-examination:
"There is no data or reasoning to support a suggestion that risk may be assigned based on any particular level of antibody titre, or what would represent a "moderate" risk of developing PML. Given the absence of reasoning and the brevity of the statement, I do not believe the Skilled Team would attach any weight to it."
- Biogen also contended that Professor Berger's cross-examination on the passage did not support Sandoz's case. They submitted that he agreed that the passage was entirely speculative and did not define any particular link between antibody level and risk, relying on this passage of cross-examination:
6 [Q.] ...coming to this today and knowing antibodies are associated
7 with risk of PML, one can read into high, moderate or low and
8 say that may or may not be teaching the same thing, but I am
9 saying if you put that information out of your mind it is not
10 clear how it is ascribing risk by reference to antibody
11 levels, is it?
12 A. Not in the given evidence at that time point.
13 Q. There is no scientific reasoning and there is no data to
14 support the teaching in this paragraph, is there? There is
15 nothing to support that antibody levels may be indicative of
16 risk; it just does not reappear in the rest of the document,
17 does it?
18 A. Definitely not in the term of this example of high, moderate
19 or low.
20 Q. It is at most a speculation. You would agree with that; yes?
21 A. As far as I read, and I am not a native speaker of course, but
22 if somebody said something in brackets "e.g.", then it offers
23 a possibility, but nevertheless I think if a neurologist, who
24 is seeking the best way to link a risk to his or her patient,
25 then of course it would be interesting to know how this may be
2 disclosed from this phrasing.
3 Q. It does not give you a reason for thinking you can assign risk
4 that way, it does not give you a reason in the sense of
5 providing data, or a reason in the sense of providing a
6 scientific argument, a hypothesis; it does not provide any
7 reason at all.
8 A. No, it does not exist at this time point.
- Accordingly, Biogen said this passage of WO369 added nothing of substance to the disclosure of Gorelik, and nothing that came close to motivating the Skilled Team to undertake a large and logistically complex study without any expectation of success, particularly when read in the context of the "bewildering" complexity of the field at the priority date.
- Dr Molyneux's written evidence was that the Skilled Neurologist would be interested to understand which of the clinical scenarios and protocols discussed in the application could prove useful as a means of assessing PML risk. When he was reminded of that, and it was suggested to him that as a result the Skilled Neurologist would be interested in seeing whether the protocol at p.6 ll.21-27 would prove useful in that regard, he took refuge in the 'single word in a long document' point.
- Furthermore, while Dr Molyneux emphasised the lack of data and reasoning, he did not suggest that the idea (or hypothesis) was one which the Skilled Neurologist would regard as one which could not be correct or was not worth looking into. This is not, therefore a case where there is a 'lion in the path' such that invention can lie simply in doing what the prior art tells you to do.
- Overall, Sandoz submitted that Dr Molyneux's suggestion that because p.6 ll.21-27 came in the middle of a long document it was not something to which the Skilled Neurologist would "pay undue heedance" was unconvincing. As I found above, it is the new disclosure of WO369. In circumstances where the ability to determine whether a patient was at high, moderate or low risk of PML would be very useful to the Skilled Neurologist, as Dr Molyneux accepted, it is hard to see why the Skilled Neurologist would not be motivated to investigate the idea.
- Further, as Dr Molyneux accepted, his evidence about the study which the Skilled Neurologist would do given Gorelik also applied given WO369. So the Skilled Team would be conducting that study anyway. Even if, which I do not accept, the Skilled Team given Gorelik would not bother to look at the nOD and % inhibition data being generated for a potential association with risk of PML, it is inconceivable that they would not do so given the clear statement on p.6 ll.21-27 of WO369.
- Sandoz submitted that Dr Molyneux had no convincing answer to that point - see T2 174/25 - 176/5:
Q. But we have this Skilled Neurologist, or Skilled Team, undertaking the study that we have talked about in the context of Gorelik, and which you said you would do the same thing given '369. So they are doing this study with lots of patients and people who go on to develop PML, and they are using an assay which gives you nOD levels; yes?
A. They are using an assay that gives you nOD levels in order to read a binary output.
Q. And they see this -- they have this statement in front of them. Why would they not look to see if this was right?
A. I believe the Skilled Neurologist reading a 35-page document would not see that single word in this document and be minded to embark on a research project or do anything other than accept it as one of a range of examples, aspects or embodiments in the document. It is a single word in a 35-page document.
Q. Doctor, you are going to be doing the study anyway, and, as we discussed before, the only difference is what data you look at. That is the difference between us. It is not whether you do the study, it is whether you look at the data in the nOD levels. What this document is saying is look at the nOD levels and correlate to risk. Why would the Skilled Neurologist, having read that, not say, "Okay, let us look at the nOD levels and see if this is right"?
A. So, my view of the Skilled Neurologist, reading the patent, is that the Skilled Neurologist would look at this and not place undue emphasis on it. You have a different view, Professor Berger has a different view, as his evidence and his report attest to.
- On this third route, on either piece of prior art, I understood Biogen to have three points in response, which I discuss in turn:
i) First, there were multiple alternative paths for the Skilled Team to explore if interested in investigating PML risk further.
ii) Second, the study which Sandoz proposed would not be routine and therefore would not be obvious to conduct, regardless of the prospect of success.
iii) Third, that there was no evidence that the Skilled Team would have been able to gain access to the large number of samples (including samples from PML patients) necessary for such a study.
- I can deal with this topic relatively succinctly. Biogen's point was that there were multiple alternative research avenues to explore. Indeed, Dr Molyneux described the debate around the pathogenesis of PML at the time as 'bewildering' and rejected any notion of a predominant theory. In cross-examination Prof. Berger was taken to a large number of papers in the L and DXX bundles where various theories on PML pathogenesis and the stochastic sequence of events required were set out. It was put that there were lots of potential matters that the Skilled Neurologist wanting to gain a better understanding of the relationship between JCV and PML would explore.
- However, as Prof. Berger pointed out, whilst that might be the case the Skilled Neurologist would "need to pick up one". As Prof. Berger said, whilst other areas would also have been obvious to explore the easiest route was the one that both he and Dr Molyneux agreed the Skilled Neurologist would be interested in, namely testing a Gorelik-type assay on a larger number of patients:
"I think the easiest way, of course, is to expand the cohort to show, and I think that was also the point that I was making before, that the meaningfulness of this paper also turns into clinical routine meaningfulness."
- I agree. The fact that there might have other obvious avenues to follows does not detract from the one suggested by Sandoz.
- This issue originated in Prof Berger's evidence in chief in this passage [Berger 1, §80]:
'The Skilled Neurologist would have wanted to review further data from a larger number of patients (both PML and non-PML) than were evaluated in Gorelik, which would have been collected as a matter of good clinical practice in any case (as I mentioned at paragraph 66 above), to determine whether the hypothesis that risk of PML would be associated with antibody levels was borne out.'
- A little more detail emerged in Prof Berger's reply report:
'12. ... If the Skilled Neurologist could not obtain nOD values for patient samples from Unilabs (or another laboratory testing samples with the STRATIFY JCV assay), conducting this investigation would involve preparing an anti-JCV antibody assay, measuring antibody levels with that assay, recording the results and analysing whether there was a correlation with PML risk. I understand from Bristows that Mr Scrimshaw's evidence is that such an assay could have been prepared by the Skilled IDS, although it would be different to that used in Gorelik. It would take time to collect the patient data but it would be straightforward to collect and analyse.'
- Biogen relied on the evidence from Dr Molyneux that an investigation into antibody levels and PML risk would constitute 'a considerable research project', but I agree that the project that Dr Molyneux had in mind in §186 of his first report was the same as Prof Berger's. Where they differed was as to (a) the data which would be recorded by the Skilled Team and (b) whether they would have access to sufficient samples.
- However, as Biogen pointed out, there was no evidence that nOD values were available from Unilabs at the priority date. Accordingly, the Skilled Team would have to perform their own data collection. In cross-examination, Professor Berger accepted that doing the study on a prospective basis was not feasible given the rarity of PML. As he put it, one could only do this '...if I have a long life...'.
- Instead, he alighted upon it being performed retrospectively. In these circumstances, Biogen said this was no longer one of a neurologist recording data for patients in the ordinary course of their clinical practice, but required a significant initiative to obtain and test samples that had previously been gathered elsewhere.
- It was clear that data gathered at one centre, even if samples were available on a retrospective basis but especially if they were only available on a prospective basis, would not be sufficient for Sandoz's case. For Sandoz's case to succeed they needed to show that samples would be available to the Skilled Team from a wide range of other sources.
- Accordingly, Biogen suggested that Sandoz's case had come down to this: that the Skilled Team, presented with Gorelik or WO369, would be motivated to obtain and perform a retrospective study on thousands of serum samples taken across multiple sites in order to investigate whether there exists any correlation between anti-JCV antibody levels and the risk of PML without any expectation that such a correlation would emerge.
- This leads me to Biogen's third point - whether the Skilled Team would have access to sufficient samples so that the pattern in the data would 'fall out', as Sandoz submitted.
- Biogen pointed out that they actually performed the clinical study required as part of their ongoing research into the safety and effectiveness of Tysabri, despite there being no expectation of success regarding the relevance of JCV antibody levels, and in doing so they made a valuable technical contribution to the art, which is used to this day in clinical practice and which Sandoz seek to make use of in their assay. On this basis, Biogen submitted the Patent involved an inventive step.
- Biogen's position was that there was no evidence that gaining access to the large number of samples (including samples from PML patients) necessary for such a study would even have been possible for the Skilled Team, still less routine. Biogen contrasted the number of samples they said was required for this study with the number required to put the teaching of the Patent into effect, although it is relevant to note that there was little exploration of how many samples would be required for either purpose. Biogen also pointed out that there had been no pleading as to the availability of such samples and the accessibility of such samples is not said to form part of the common general knowledge.
- Professor Berger suggested in cross-examination that such samples might come either from Biogen or a larger MS centre or consortium [T5/6464-15]. As to the first option, Biogen submitted that the Skilled Team cannot be deemed to have had access to Biogen's samples as a matter of law. On the facts, Professor Berger was himself working in cooperation with Biogen around the priority date. Biogen suggested this had coloured his view. Overall, I agree that there was no evidence that Biogen made samples available for testing by others.
- Biogen also suggested that there was no evidence that obtaining and testing samples from a larger MS centre or consortium would have been possible for the Skilled Team, still less routine. Biogen submitted there was evidence of only one such study being done, reported in the Trampe paper (which emerged for the first time in Dr Molyneux's cross-examination materials). However, as Dr Molyneux pointed out, there were indications that the idea for this study originated from Biogen and might have been made possible by Biogen's support.
- I can now state my findings on this aspect of the case. The case of obviousness over WO369 is the stronger one and its teaching would give the Skilled Team a strong motivation to investigate further. So, there is no problem with motivation.
- Although, as I indicated, there was little exploration of how many samples would be required in the study before the association between anti-JCV antibody levels and the risk of PML would emerge as those data were collected, the data presented in the Patent provide an indication. Furthermore, as mentioned above, Prof Berger indicated the Skilled Team would want to evaluate a larger number of patients (both PML and non-PML) than were evaluated in Gorelik. I suspect that Dr Molyneux was exaggerating somewhat when he indicated 'thousands' would be required.
- Perhaps because this third route appears to have been something of an afterthought in Sandoz's evidence, the evidence that a sufficient number of samples would have been available to the Skilled Team was thin. Whilst one would hope that MS centres would wish to cooperate in such a study, I regret to say that, on balance, I was not persuaded that Sandoz had proved that a sufficient number of samples would have been available to the Skilled Team within any reasonable time period. Whilst there is a degree of legal fiction in the concept of the notional Skilled Team doing work at the priority date having read each piece of prior art, I do not consider this can be stretched so as to allow the Skilled Team years to accumulate samples or time to negotiate access to samples from particular centres.
- Had the samples been available, I agree that the conduct of the study would have occurred and would have been routine work for the Skilled Team to collect and analyse the data. With sufficient data, the Skilled Team would have been able to define their own dividing line between high and low risk of developing PML.
- As should be clear from the conclusion just stated, even if I am wrong about sufficient samples being available to the Skilled Team, the end point would not have rendered claim 1 obvious on either of Constructions 5 or 6, precisely for the reasons which underpin the insufficiency case. On Construction 4, it would have been a matter of happenstance that the population studied by the Skilled Team hit the median of antibody positive patients, so the claim would not have been obvious on Construction 4. For these reasons and based on the evidence presented, I am unable to find the Patent obvious on any of those Constructions.
- That leaves Constructions 1, 2 and 3. On any of these Constructions, WO369 disclosed having a dividing line between seropositive patients at higher and lower risk of developing PML based on the level of anti-JCV antibodies. Necessarily, the level of anti-JCV antibodies at this dividing line would be expressed as an index value. On this basis, if any of Constructions 1, 2 or 3 was correct, I would be inclined to find that claim 1 made no technical contribution to the art. In case it is not apparent, my conclusion on Constructions 1, 2 & 3 does not depend on sufficient samples being available to the Skilled Team.
- Sandoz's Confidential Re-Amended PPD ("the PPD" [F2/1]) sets out the nature and components of the relevant aspect of the Sandoz Assay and how it is used (see §§6-16) and annexes the proposed instructions for use of the assay (Annex 4). It also annexes the Summary of Product Characteristics associated with the Tyruko product in Great Britain, the Risk Management Plan and EU Physician Information and Management Guidelines for the Tyruko product (Annex 1, Highly Confidential Annex 2 and Annex 3), and sets out pertinent aspects of those documents at §§22-24A. The PPD also sets out information relating to the correlation between index values from the Sandoz Assay and those from the Biogen STRATIFY JCV DxSelect assay - see §§17-20. Finally it explains at §25 that testing of samples from patients in the UK will be performed outside the UK by a third party (Medicover) and provides the draft JCV Portal User Guideline which explains the proposed logistics for testing for patients in the UK (Highly Confidential Annex 5), also addressed by Mr Will in his third and fourth witness statements. Since this issue may engage more than just the Sandoz Assay, I will refer to the whole process as the 'Sandoz Method'.
- For present purposes, the following summary of the Sandoz Method should suffice:
i) Having registered on the Medicover JCV Portal and activated their account, the clinician orders the relevant materials and registers an individual patient.
ii) The clinician places an order for JCV testing and schedules a pickup of the patient sample.
iii) Using the supplied materials, the clinician takes a blood sample from the patient and packages it for collection by courier.
iv) The blood sample is taken by courier overseas to Medicover.
v) The assay is conducted outside the UK by Medicover.
vi) When the test results are ready, the clinician receives an email notification. The clinician then logs onto the Medicover JCV Portal and can view and download the report of the results.
- During my pre-reading, I noted that in the draft JCV Portal User Guidelines in Annex 5, the result was shown as merely 'positive', so in opening I raised the question of what the clinician will actually see reported as the result from the Sandoz Assay. However, Sandoz had already pleaded that the clinician will access the test results of the assay, which results include the anti-JCV antibody index value, and another version of the typical test result page showed the index value will be stated to 2 decimal places (in the example, 0.04).
- Sandoz pleaded that:
'Clinicians in the UK will do no more than interact with the patient's body to obtain a sample for diagnostic purposes and/or conduct the intellectual exercise of using the results of the Assay to determine whether a patient's anti-JCV antibody status is such that treatment with natalizumab is appropriate.'
- Biogen pleaded that Sandoz threatens and intends to, in the UK: (i) use the method of the claims of the Patent; and/or (ii) offer the method of the claims for use. Biogen did not plead explicitly that Sandoz threatens and intends to offer the method of the claims for use in the UK, but Sandoz accepts that must be implicit, as it is a necessary part of an allegation of infringement pursuant to section 60(1)(b) of the Patents Act 1977.
- While Biogen pleaded that Sandoz itself threatens and intends to use the method of the claims in the UK, as Sandoz submitted, it appears that its real allegation is that clinicians will be using the method of the claims in the UK and that Sandoz (itself or in common design with Medicover) will be joint tortfeasors with the clinicians.
- Similarly, the allegation of offering for use is that Sandoz (itself or in common design with Medicover) is offering the method of the claims for use by clinicians in the UK.
- The matters relied upon by Biogen in support of its contention that the claimed method will be used by, and offered for use by, clinicians in the UK, are (i) that patients will have blood samples taken in the UK and will be treated with Tyruko in the UK, and (ii) that clinicians in the UK will make use of the results of the Sandoz Assay as part of an assessment of whether a patient in the UK is at increased risk of developing PML.
- Sandoz contends that these matters do not mean that clinicians in the UK are using the method of the claims (or that Sandoz is offering the method for their use in the UK). The method of claim 1 comprises determining an anti-JCV antibody titer in a patient serum or plasma sample by an ELISA assay. That ELISA assay involves the steps of adding an aliquot of the sample to a substrate bearing HPVLPs and detecting the level of anti-JCV antibody bound to that substrate. Further, the anti-JCV antibody titer must then be expressed as an index value by normalising the OD of the sample to that of the calibrator. All of those steps are carried out outside the UK.
- As noted above, Biogen relies on the fact that the blood samples are taken in the UK, and that the patient is treated with natalizumab in the UK (if at all). But those matters are not features of the claims. The only relevant matter that takes place in the UK is that the clinician uses the index value generated by the assay as part of their clinical assessment of risk of PML.
- My attention was drawn to the following passages in the authorities, which are well-known but nonetheless repay careful reading.
- In Menashe v William Hill [2002] EWCA Civ 1702 claim 1, so far as relevant, was to:
"a gaming system for playing an interactive casino game comprising a host computer, at least one terminal computer forming a player station, communication means for connecting the terminal computer to the host computer and the program means for operating the terminal computer, the host computer and the communication means ... characterised in that the terminal computer is situated at a location remote from the host computer ...".
- The allegation was of infringement under s.60(2) by the supply to punters in the UK of "a program usually by a CD which turns the punter's computer into a terminal computer which communicates via the net with a host computer [which] has the properties and carries out the functions of a host computer referred to in the claims, but...is located outside the United Kingdom".
- Jacob J had held that it was enough for infringement under s.60(2) if the supply of the means in question had an effect in the UK. The Court of Appeal rejected that. Aldous LJ held that infringement under s.60(2) required the invention to be put into a state of effectiveness in the UK, i.e. that the means must put the claimed apparatus into an infringing state in the UK - see [24]-[29].
- Nevertheless, in that case there was infringement for the reasons explained in [33]:
"If the host computer is situated in Antigua and the terminal computer is in the United Kingdom, it is pertinent to ask who uses the claimed gaming system. The answer must be the punter. Where does he use it? There can be no doubt that he uses his terminal in the United Kingdom and it is not a misuse of language to say that he uses the host computer in the United Kingdom. It is the input to and output of the host computer that is important to the punter and in a real sense the punter uses the host computer in the United Kingdom even though it is situated in Antigua and operates in Antigua. In those circumstances it is not straining the word "use" to conclude that the United Kingdom punter will use the claimed gaming system in the United Kingdom, even if the host computer is situated in, say, Antigua. Thus the supply of the CD in the United Kingdom to the United Kingdom punter will be intended to put the invention into effect in the United Kingdom."
- Thus, the system as a whole was used by the punter and that use took place in the UK, even if part of the system was outside of the UK.
- Menashe may be contrasted with the facts of RIM v Motorola [2010] EWHC 118 (Pat). In RIM v Motorola, the claim was to a messaging gateway system. The claimed system included the user's wireless devices and a messaging gateway. RIM's servers (the messaging gateway) were in Canada, but emails were sent by and to users in the UK. Arnold J considered Menashe at 155, quoting the passage from Aldous LJ at [33] and continuing in 156:
"156. I agree with RIM that asking and answering Aldous LJ's questions in this case leads to a different answer. Who uses the method of operating a messaging gateway system that has the claimed features? The answer is RIM. Where do they operate it? The answer is in Canada."
- So Arnold J held that the claimed method was not offered for use in the UK, nor was there infringement under s.60(2), because RIM was using the claimed method in Canada and not in the UK. The fact that subscribers with devices in the UK could use the messaging gateway system to communicate with a remote messaging system located in the UK did not lead to infringement.
- The principle established in Menashe was applied in the context of s.60(1)(b) in Illumina v. Premaitha [2017] EWHC 2930 (Pat) at [502] - [508] per Henry Carr J.
- To understand that case, it is necessary to delve somewhat into the technical details. In Illumina one of the claims in issue was to:
"A detection method performed on a maternal serum or plasma sample from a pregnant female, which method comprises detecting the presence of a nucleic acid of foetal origin in the sample, wherein said nucleic acid is a paternally inherited sequence which is not possessed by said pregnant female."
- The other claims in issue were more complicated. One read as follows:
"A method for performing prenatal diagnosis of a foetal chromosomal aneuploidy in a biological sample obtained from a female subject pregnant with a foetus, wherein the biological sample is maternal plasma or serum and wherein the sample includes cell-free nucleic acid molecules from the female subject and the foetus, the method comprising:
performing a random sequencing on at least a portion of a plurality of the nucleic acid molecules contained in the biological sample to obtain a pre-determined number of sequences, wherein the sequences represent a fraction of the human genome;
aligning, with a computer system, each sequence to a human genome;
determining a first amount of sequences identified as being aligned to a first chromosome;
determining a second amount of sequences identified as being aligned to one or more second chromosomes;
determining a parameter from the first amount and the second amount; wherein the parameter represents a relative amount between the first and second amounts; and
comparing the parameter to one or more cut-off values, to determine a classification of whether a foetal chromosomal aneuploidy exists for the first chromosome."
- Henry Carr J had to consider whether these claims were infringed by Premaitha's Alternative Proposed Process in which it proposed moving certain automated computerised steps off-shore (see [500]):
"In summary, the DNA preparation and sequencing is (to be) conducted in the UK but the multiplexed raw i.e. unmapped sequence data resulting from those steps, along with patient and sample data, is then sent to a data analysis site in Taiwan where the remainder of the steps are carried out. A PDF format test report is then returned to the UK. Specifically, the Alternative Proposed Process comprises the following steps:
i) receiving a blood sample from a patient in the UK;
ii) carrying out the preparatory steps and the sequencing processes in the UK;
iii) sending the raw data comprising the results of the sequencing reads electronically to Taiwan;
iv) performing the analysis of the data in Taiwan, including the Rx calculation, sex determination and foetal fraction estimation;
v) generating a report in Taiwan;
vi) sending the report back to the UK;
vii) receiving and unpacking the report in the UK and formatting it for printing, storage and sharing with the patient.
- Henry Carr J explained his reasoning at [507]-[508]:
"In my judgment, the crucial question is where, in substance, is the Alternative Proposed Process to be used? The answer is the United Kingdom. In substance, the 'method of detecting a nucleic acid of foetal origin in a sample' (Lo 1), the 'method of detection of foetal aneuploidy' (Quake) and the 'method for performing prenatal diagnosis' (Lo 2/3) would be performed by laboratories in the UK. The blood test would be taken in the UK, the sequencing machine would be operated in the UK and the information so obtained would be transmitted to Taiwan for a pre-determined set of automated computer processes to be applied to it. The output of the computer processing would be sent back to the United Kingdom for use in the United Kingdom.
As Aldous LJ said in Menashe, it does not matter where the computer is situated. The process is operated, in substance in the UK. I accept Illumina's submission that any other result would make it far too easy to avoid infringement of patents of this nature, given the ease of digital transmission and the ability to off-shore computer processing."
- So in that case in substance the methods of the claims were being used in the UK. All that had been taken off-shore was the automated computerised aspects of the process, which conducted the analysis of the raw data which had been obtained in the UK.
- Applying those principles, Sandoz submitted as follows:
i) If one asks where in substance the method of the claims of the Patent is to be used, the answer must be that it is outside the UK.
ii) The core of the method is the use of the ELISA to analyse the patient sample and generate the index value. All of that is carried out outside the UK.
iii) All that happens in the UK is that the clinician uses the index value that has been obtained from the ELISA abroad as part of their clinical assessment in order to advise a patient of their risk of PML. That is a non-technical aspect of the claims and can hardly be said to be the substance of the claimed method.
- For their part, Biogen submitted that Sandoz's territorial defence to infringement was bad in both fact and law. Biogen characterised Sandoz's case as being based on one of two propositions: either (i) the user of the Sandoz Method is the lab that performs the Sandoz Assay and not the clinician, or (ii) the lab is one of the users of the Sandoz Method, and all users must be in the UK for the method to infringe under s.60(1)(b).
- Biogen submitted that proposition (ii) was bad in law, there being no authority to support it.
- On the facts, Biogen submitted the first question to be asked is who uses the Sandoz Method? Here, they submitted, the answer is plainly the clinician (with the result that proposition (i) was untenable). It is they that procure the sample of serum, initiate testing of that sample and use the result in the evaluation of risk. The lab that accepts and processes the sample merely generate data for use by the clinician. In those circumstances, to paraphrase Aldous LJ, it is of no relevance to the clinician, nor the patentee, whether or not the lab is situated in the United Kingdom.
- There is no doubt that step (i) of the method, the determining of the anti-JCV antibody titer, is carried out outside of the jurisdiction. Although the blood sample is taken in the jurisdiction, there is no doubt that the serum or plasma sample for testing is prepared at Medicover, outside the jurisdiction along with the performance of all the other steps in the assay. As for step (ii), it involves two determinations:
i) The first is the determination of the resulting index value. That determination also takes place outside the jurisdiction.
ii) The second is the determination that the patient is at high risk of developing PML if the anti-JCV antibody index value is determined to be >1.5.
- In order to decide where the second determination takes place, it is necessary to understand what this determination is. The whole point of this method, as I understand matters, is that it is deterministic i.e. if the index value is >1.5, then it is determined the patient is at high risk of developing PML. No exercise of clinical judgment is required. Of course, based on the actual index value, the clinician may go on to make his or her own assessment of the clinical risk, but that assessment is not part of the claimed method.
- On this basis, it seems entirely artificial to sever the second determination from the first. Therefore, the second determination also takes place outside the jurisdiction.
- One can test this conclusion by considering two hypothetical scenarios:
i) First, where the clinician happens to be visiting Medicover and is provided with the results for his patient.
ii) Second, where the clinician happens to be abroad (either on holiday or at a conference) and logs onto the Medicover portal to see his or her patient's results.
In both cases, the determination in step (ii) has been made because it is built into what occurs at Medicover.
- However, I should consider whether the fact that the final step in the overall process is that the index value is reported to the clinician in the UK makes any difference. It is certainly the case that the clinician and his or her patient receive the benefit of the index value and the determination whether the patient is or is not at high risk of developing PML i.e. it is useful to them. That, however, does not mean that Sandoz/Medicover/the clinician is using the method of the claim in the UK. It is safe to assume they use the result or the outcome of the method in the UK (whether the determination or not of high risk, or the index value itself), but, in my judgment, the method specified in the claim is not used in the UK.
- In case I am wrong in this conclusion I will determine the technical issues which arise.
- On the facts, the issues involve allegations of infringement both on a normal construction and on the basis of equivalence. I should remind myself of the relevant legal principles.
- The Actavis questions are:
(1) Notwithstanding that it is not within the literal (that is to say, I interpolate, normal) meaning of the relevant claim(s) of the patent, does the variant achieve substantially the same result in substantially the same way as the invention, i.e. the inventive concept revealed by the patent?
(2) Would it be obvious to the person skilled in the art, reading the patent at the priority date, but knowing that the variant achieves substantially the same result as the invention, that it does so in substantially the same way as the invention?
(3) Would such a reader of the patent have concluded that the patentee nonetheless intended that strict compliance with the literal meaning of the relevant claim(s) of the patent was an essential requirement of the invention?
- As Sandoz submitted, before the Court can answer the Actavis questions, it must identify the "inventive concept", explained by Kitchin LJ in Icescape Ltd v Ice-World International BV [2018] EWCA Civ 2219 as "the inventive core". However, as observed by Nugee J in E Mishan & Sons Inc v Hozelock Ltd [2019] EWHC 991 at [83], the question arises as to the level of generality at which the inventive concept should be stated. One can often state an inventive concept at a level of generality such that it is adopted by the defendant, but that may lose all contact with the language of the claim. It must be remembered that under the Protocol to Art 69 EPC, the claim must not be treated merely as a guideline so that protection can extend to what, from a consideration of the description and drawings, the patentee has contemplated. As Floyd LJ said in Icescape at [97]:
"It should not be thought, however, that the claims do not continue to have an important function. It is variants from the claim which have to achieve substantially the same effect in substantially same way as the invention. The claims remain the starting point for the subsequent analysis of variants. Although we may have edged closer to it, the new approach does not transgress the second of the outlawed approaches in the Protocol, which treats the claim merely as a somewhat vague guideline."
- The approach adopted by Arnold J in Akebia Therapeutics Inc v Fibrogen Inc [2020] EWHC 886 was therefore to ask what the inventive concept embodied in the claim was - see e.g. [419]-[421] (see also Mishan at [84]). Similarly, in Kwikbolt Ltd v Airbus Operations Ltd [2021] EWHC 732 (IPEC), HHJ Hacon explained at [94] that: "the inventive concept is the new technical insight revealed by the invention as claimed, in the context of the specification as a whole, as it would have been perceived by the skilled person at the priority date." Sandoz accordingly submitted that to focus on the inventive core of the claim, or the inventive concept embodied in the claim, is the right approach, because the purpose is to identify those parts of a claim which matter to its inventive concept, and those which are immaterial to it. Thus, in Actavis itself the use of the sodium cation was immaterial, just as in Icescape, claim integers D and E were part of the common general knowledge and so immaterial to the inventive core of the claim.
- Sandoz were keen to stress the above points because they emphasise the importance of properly identifying the inventive concept underlying the claim. They acknowledged that a patentee is entitled to rely upon the doctrine of equivalents to broaden out their claim to catch alleged infringements that take the inventive concept of the claim even if not all integers of the claim are taken. The doctrine applies where what is taken is the 'inventive core' of the claim. On the other hand, Sandoz submitted that what the doctrine of equivalents does not allow a patentee to do is effectively re-write the claim by broadening out the inventive concept of the claim to something at a higher level of generality which may be contemplated by the description. Sandoz submitted that was precisely what Biogen was seeking to do.
- The final point which Sandoz sought to make about Actavis question 3 is that it acts as a limit on what the patentee can do. In particular, the courts have recognised the "disclosed, not claimed" principle, namely that where a patentee sets out a number of ways in which the invention can be implemented in the patent but then uses language to limit the claim to just one implementation, the patentee cannot then rely upon the doctrine of equivalents to broaden out again. This was explained by HHJ Hacon in Philip Morris v Nicoventures [2023] EWHC 2616 (Pat):
89. Since the Actavis judgment courts at first instance have had regard to a principle well established in the German Federal Court of Justice, considered and endorsed by Arnold J in Akebia Therapeutics Inc v Fibrogen Inc [2020] EWHC 866 (Pat), at [454]:
"If the description discloses a plurality of possibilities for achieving a specific technical effect, but only one of those possibilities is catered for in the patent claim, the utilisation of any other possibilities properly does not constitute infringement of the patent with equivalent means."
90. This "disclosed but not claimed" principle has since been relied on at least twice, see Facebook Ireland Limited v Voxer IP LLC [2021] EWHC 1377 (Pat) at [201] and Shenzhen Carku Technology Co., Ltd v The Noco Company [2022] EWHC 2034 (Pat) at [106]-[112].
- In order to understand the technical issues which arise over the index value, it is necessary to outline some more detail about the Sandoz Assay. For entirely understandable reasons, the Sandoz Assay was designed to be as close as possible to the STRATIFY JCV DxSelect assay currently marketed by Biogen. If a patient has an index value of >1.5 from the STRATIFY JCV DxSelect assay, then he or she is determined to be a high risk of PML.
- In order to obtain approval from the EMA, Sandoz carried out a correlation of index values obtained from the Sandoz Assay vs the STRATIFY JCV DxSelect assay. It was on the basis of that correlation that the EMA decided that the index value from the Sandoz Assay which should be used as the start of the higher risk range should be 1.4. Therefore, clinicians following the guidance in the regulatory documents will treat patients with an index value of 1.4 or above as being at increased risk of PML.
- An additional complication is that the detailed data underpinning the correlation indicates that an index value of 1.5 in the STRATIFY JCV DxSelect assay corresponds to an index value of 1.451 in the Sandoz Assay. That, it seems to me, does not detract from the fact that a patient with an index value of >1.4 from the Sandoz Assay is effectively determined to be a high risk of PML and will be treated accordingly.
- Biogen appeared to take a number of points:
i) First, they said it was not clear that >1.4 will be the cut-off, relying on the 1.451 value which corresponds to 1.5 in the STRATIFY JCV DxSelect assay.
ii) Second, Biogen say that using a cut-off of >1.4 does not take the Sandoz Assay outside the claim because the >1.5 feature must be construed as covering any value >1.45.
iii) Third, Biogen also point out that there will be a very large degree of overlap between patients whose Sandoz Assay index values are >1.5 and those whose index values are >1.4. Biogen invited consideration of Figure 5 in the PPD, and imagining horizontal lines originating at 1.4 and 1.5 on the Y-axis. The difference, they submit, in terms of subdivision of the total patient population, is minimal. Accordingly, on a purposive construction, Biogen submits that the Skilled Team would understand a cut off of 1.4 to be close enough to 1.5 to be within the scope of the claim.
- Sandoz understood the overlap point differently: that Biogen was contending that because there is an overlap of patients (assume substantial) determined by both assays to be high risk, that is sufficient for infringement.
- I can deal briefly with each of Biogen's points:
i) I consider the point is clear, since the EMA has decided that >1.4 is the cut-off for the Sandoz Assay.
ii) The second point I have determined under construction above. In any event, as Sandoz submitted, the debate as to whether ">1.5" would be understood as meaning, in fact, "> (1.45 to 1.54)" is an arid one. The Sandoz Assay uses a cut-off of 1.4 as denoting high risk and therefore falls outside the range 1.45-1.54. The fact that 1.5 in the STRATIFY assay is equivalent to 1.451 in the Sandoz Assay is irrelevant for these purposes but, in any event, it makes Biogen's position worse and not better because the cut-off applied for the Sandoz Assay is 1.4.
iii) Both formulations of the overlap point are at entirely the wrong level of generality demanded by the claim. The claim determines whether an individual patient is at high risk of PML. According to the claimed method, an individual patient is either at high risk or not, depending on a comparison between their index value and the relevant limit. The relevant limit for the Sandoz Assay is >1.4.
- Accordingly, I find the Sandoz Assay would not infringe on a normal Construction 5.
- On Construction 6, there is a further point against infringement. Whilst it is clear that Sandoz tried to make their assay equivalent to the STRATIFY JCV DxSelect assay, as Sandoz pointed out, Biogen have produced no evidence to demonstrate that the Gen2 assay used in the Patent is the same as the STRATIFY JCV DxSelect assay.
- On Construction 4, I received no argument to the effect that the cut point in the Sandoz Assay is positioned at/around the median of the relevant population in the UK, although I suspect it is.
- Although it is counterintuitive to apply the concept of 'infringement on a normal construction' to Constructions 1, 2 and 3 (which by no stretch of the imagination are 'normal' constructions) the Sandoz Assay would infringe if any of those constructions were correct.
- Even if Sandoz do not meet the >1.5 feature on a normal construction (i.e. Construction 5), Biogen say that the Sandoz Assay is an immaterial variant which infringes under the doctrine of equivalents. For the purpose of this argument, Biogen appeared to be relying on a combination of their inventive core and median arguments. It was put this way in their written closing:
'Anti-JCV antibody titre must be determined by means of an ELISA assay that uses HPVLPs as the capture antigen. This must be expressed as an index value obtained by normalising to a cut-off calibrator made with a mix of anti-JCV antibody positive and negative serum (fn:The ELISA must also use positive and negative controls that comprise positive and/or negative serum and have defined index values, but nothing discrete turns on these). The patient is then determined to be at high risk of developing PML if that index value is > 1.5. This value derives from the teaching that, when the Gen2 assay is used, an index value of > 1.5 covers approximately the upper 50% of all anti-JCV antibody positive index values but 87% of PML cases (see Example 6).
While the claim requires anti-JCV antibody titres associated with a high risk of PML to be expressed as index values >1.5, the use of that index value as such to identify the cut point forms no part of the inventive concept of the claim. The same cut-point could be expressed by a different index value simply by adjusting the dilution of the calibrator. To use the technical contribution of the Patent it is sufficient to use an index value to represent a clinically relevant cut point to distinguish patients at greater risk of developing PML. That is the inventive concept or core of the claim.'
- On that formulation of the inventive core, Biogen submitted that the Actavis questions must be answered yes, yes and no for both potential variants, individually and in combination. I will deal with the index value first.
- As the authorities indicate, the Actavis analysis (particularly for question 3) is dependent on a correct identification of the inventive concept. Furthermore, the authorities are clear that the inventive concept must be formulated at the level of generality of the claim.
- It is clear that in putting forward Constructions 1, 2 and 3, Biogen are treating the claim as a vague guideline, notwithstanding the content of the specification. So I am confident none of them represent the correct inventive concept. For different reasons, the same conclusion applies to Construction 4, which is founded on concepts which are not mentioned in the claim anywhere.
- Sandoz's suggested inventive concept was based on Construction 6 and reads as follows:
'....the inventive concept of the claim is a method of assessing PML risk by determining whether the patient has a level of anti-JCV antibodies which is above that represented by an index value of 1.5 using the Gen2 assay of the Patent.'
- This formulation is incorrect, because it depends on having already written into the claim all the details of the Gen2 assay.
- For the purposes of Construction 5, this is a claim which has a number of specific requirements for the assay. It is therefore difficult to encapsulate these in a summary without raising the level of generality significantly and impermissibly above that of the claim. To illustrate this, the patentee could have claimed an ELISA at a much higher level of generality, in which case the inventive concept could have been formulated as an ELISA for a stated purpose. That is not how the patentee chose to formulate the claim. For these reasons, I must proceed on the basis of the claim wording, albeit I should recognise those parts which have greater significance to the invention, and inevitably attention focusses on the 1.5 index value.
- However, it is important to assess the significance of the 1.5 cut point carefully, because it is all too easy to proceed on the implicit assumption that the 1.5 cut point is meaningful because of its applicability in the Gen2 assay.
- On the one hand, having considered the specification, the Skilled Team would understand that the patentee had selected the 1.5 cut point on an empirical basis and as the best cut point for the purpose of dividing between low and high risk of developing PML. This 1.5 cut point is the ultimate conclusion of all the experimental work set out in the specification, and is therefore critical to the claimed invention. On the other hand, the Skilled Team would also understand that that 1.5 cut point has that significance only when applied in the context of the Gen2 assay or an equivalent. Since the Gen2 assay is not part of the claim, and, as I have indicated, on Construction 5 the claim covers a class of assays, the significance of 1.5 becomes much more elusive.
- Due to the fact that Construction 5 only emerged in the course of the trial, there was almost no exploration of the scope of the claim on that construction. Doing the best I can, there is no doubt that the class would include a number of assays which did essentially the same job as the Gen2 assay and resulted in a meaningful division between low and risk of developing PML. One can envisage that slight changes to some of the measures specified in the claim would not make any material difference. However, I consider the class would include some assays which would either not prove useful to divide between low and high risk or would put the dividing line in a very different place to that of 1.5 in the Gen2 Assay. This is, effectively, a breadth of claim point.
- So the 1.5 cut point only has significance in the context of the Gen2 assay or equivalent.
- Although, as I have indicated, a formulation of the inventive concept is a difficult exercise, I must address the Actavis questions:
i) In my judgment, if one works on the basis that the 1.5 cut point has significance in the context of the Gen2 assay or equivalent, then it is clear that the variant achieves substantially the same result in substantially the same way as the invention.
ii) That would be obvious to the person skilled in the art, notwithstanding their realisation that the claim covered a class of assays, not all useful.
- As for Actavis question 3, Sandoz argued that this was a case of 'disclosed but not claimed' and so the Skilled Team would conclude that strict compliance with the literal meaning of the claim was an essential requirement. They point out that different index values are identified throughout the Patent as denoting high or higher risk, in particular Example 4 (>1.0 = higher risk), Example 6 (>1.5 = higher risk); Table 10 (various index values may denote higher risk), [0106] & [0108] (>3 = higher risk). Yet, when it comes to claim 1, an index value level of 1.5 is specified. Given the disclosure elsewhere in the Patent, whatever the Skilled Team understands the various index values disclosed in the Patent to signify - specific antibody levels as identified in a specific assay (e.g. Gen1 or Gen2) or, as Biogen appears to suggest, just labels with no technical significance - they recognise that the patentee has specified '1.5' in claim 1. Therefore, so Sandoz submit, only use of an index value >1.5 to denote high risk will do.
- I am not persuaded by these arguments, for a number of reasons:
i) First, one has to be careful with the 'disclosed but not claimed' argument. As an example, consider the pemetrexed case where the potassium or other cations had been mentioned in the specification. I question whether that would have made the difference to the result. Of greater importance is the significance of the feature in question to the invention as a whole.
ii) Second, it is important to take account of two features: first, the empirical nature of the 1.5 cut point and second, the relative nature of risk. The Skilled Team would realise that other values around 1.5 could easily have been chosen as the dividing line between low and high risk. This demonstrates one can take the whole benefit of the invention with a value other than 1.5.
iii) Third, the Skilled Team would also be well aware that by tweaking the set up of their assay in immaterial ways, they could engineer a cut-point at a value of 1.4 or 1.6 and still divide the same population in the same way as the Patent.
- However, it should be noted that this conclusion is dependent on attaching significance to the 1.5 cut point, which only occurs in the context of the Gen2 assay or equivalent. If that significance is missing, then, in my view, the opposite conclusion would be reached. Accordingly, this finding is intimately tied up with the decisions on construction and insufficiency.
- Claim 1 requires that the cut-off calibrator and positive control "comprise a mixture of serum positive for anti-JCV antibodies and serum negative for anti-JCV antibodies". Sandoz submit that, on a normal construction, this requirement means that they must be made from a mixture of antibody positive and negative serum.
- The Sandoz Assay uses phosphate buffered saline (PBS) diluent and bovine serum albumin (BSA) carrier protein (together with sodium azide as a preservative) as the diluent for its positive control and cut-off calibrator [PPD §8].
- Biogen did not allege that this buffer is 'antibody negative serum' within the meaning of the claims. Biogen's point was that, as a diluent, it performs precisely the same function as using serum negative for anti-JCV antibodies.
- Biogen's point was that there was no material difference between a sample (calibrator or control) that has been diluted to the right concentration with buffer and a sample which has been diluted with serum, because the resulting control or calibrator is then diluted a hundred-fold with buffer. Indeed, the difference in the amount of buffer in each case is within the pipetting margin of error. The cross-examination on this topic was at day 4, page 538, line 20 - 542, line 6, the witness accepting it made "very little" difference. In fact it is unclear how much buffer is added to the calibrator. It is prediluted, so whereas Biogen inferred it will be 100 x dilution, that was not stated in the PPD. Biogen suggested that in so far as the PPD is missing relevant information, indulgences should not be given to Sandoz.
- Biogen argued that, on a purposive construction, the Skilled Team would not distinguish it from serum negative for antibodies. This argument confuses an equivalents analysis with purposive construction. The Sandoz Assay does not infringe on a normal construction.
- Sandoz did not argue for non-infringement on the basis of Actavis questions 1 and 2. However, they submitted that infringement is precluded by Actavis question 3, for the following reasons.
- First, it is common ground that diluents other than negative serum were well known and commonly used in ELISAs at the priority date. Indeed, the Patent identifies such diluents in its general disclosure at paragraph [0129]:
"... Diluents can include, in non-limiting examples, solutions that include BSA, phosphate buffered saline (PBS), or PBS containing Tween."
- Second, despite the state of the common general knowledge and despite its disclosure of PBS/BSA diluents, the patentee goes on to claim a specific diluent, i.e. serum negative for anti-JCV antibodies. Accordingly, the Skilled Team would conclude that the patentee had decided to exclude the use of other diluents and limit itself to the use of "serum negative for anti-JCV antibodies" to achieve its aims.
- Biogen accepted that the option of using PBS/BSA to dilute controls and/or calibrators instead of antibody negative serum was CGK. Biogen characterized Sandoz's case as dependent on the claim being to an "optimized analytically validated, sensitive assay for detecting the presence of JCV antibodies in serum or plasma, i.e. a finished commercial ELISA.", a proposition which Sandoz denied. Biogen's point was the claim is to a method of evaluating the risk of PML that employs an ELISA whose function is to measure anti-JCV antibody titre, and this is described in general terms, whereas Sandoz submitted the claim is to a method which involves the use of an assay suitable for making a clinical decision.
- Biogen accused Sandoz of confusing functional equivalence with the changes that can and cannot be made to a finished, commercial ELISA. They are not the same thing.
- However, Sandoz appeared to agree with Biogen's characterisation of the claim. Their key point was this is an instance of 'disclosed but not claimed'.
- I did not understand Sandoz to dispute Biogen's point that the diluent used in the Sandoz Assay performs precisely the same function as negative serum. I do not think the 'disclosed but not claimed' argument saves Sandoz on this issue. This proposition can prove too much. If it is applied across the board then it would eliminate a large part of the doctrine of equivalents. I conclude that the Skilled Team would not conclude that strict compliance with the language of the claim on this point was an essential requirement.
- Accordingly, on Construction 5, I conclude that the Sandoz Assay would infringe claim 1 on the basis of equivalents, if the claim was valid.
- Sandoz drew attention to a pending divisional application deriving from the PCT (EP 4,187,248 A1) (the "New Divisional"). In those circumstances, Sandoz seek the following declaration as a means of obtaining '...commercial certainty as to whether their path is clear to supply the Product and/or the Assay...'. Sandoz characterised this as Arrow relief, a point hotly disputed by Biogen:
"... as at any date between 31 May 2011 and 31 May 2012 inclusive, the PCT did not disclose an in vitro JCV assay clearly enough and completely enough for a person skilled in the art to perform such assay to determine the anti-JCV antibody titer of a serum or plasma sample from a patient expressed as an index value (nOD) and, from that index value, to determine the level of risk of that patient of developing PML wherein high risk of developing PML corresponds to an index value of >1.5 or wherein a particular risk of developing PML corresponds to any other index value disclosed in the PCT."
- There was no real dispute as to the state of the authorities on Arrow relief. Biogen accepted (in a slightly grudging fashion) that in a 'handful' of cases where pending divisionals have created prolonged legal uncertainty over a particular product, the Court has granted a so-called Arrow declaration. The nature of such a declaration was explained by Floyd LJ in Glaxo v. Vectura [2019] RPC 2:
"14. The Arrow declaration is, in effect, a declaration that a party has a Gillette defence as of a particular date against attacks by later patents. The Gillette defence can be traced to the speech by Lord Moulton in Gillette Safety Razor Co v Anglo- American Trading Co Ltd (1913) 30 R.P.C. 465. In a Gillette defence a defendant contends that the product he is selling was obvious at a particular date, and cannot accordingly fall within a valid claim of a later patent. Although such a defence is raised in circumstances where the defendant is sued on specific patents, there is no reason why a properly worded declaration that a product is obvious at a particular date cannot provide protection against any later patent. As pointed out in the Arrow case itself, however, in order to render the issues for the court properly justiciable, the characteristics of the product in respect of which the declaration is sought must be clearly defined (see per Kitchin J. at [40], [59])".
- Floyd LJ continued:
"15. In Fujifilm Kyowa Kirin Biologics Co., Ltd. v AbbVie Biotechnology Limited and another [2017] EWCA Civ 1, [2017] RPC 9 ['Fujifilm'] this court examined for the first time whether Arrow declarations were available in principle, or whether there existed fundamental obstacles to their grant. Thus it was contended on behalf of the patentee inter alia that:
(i) an Arrow declaration was in effect a challenge to validity of a patent which could only be made once a patent was granted in the proceedings identified in s.74 of the Patents Act 1977;
(ii) such declarations were in substance a collateral attack on proceedings within the EPO, which the English court would not permit;
(iii) to allow declarations in the Arrow form would be to open the floodgates;
(iv) if the Arrow declaration does raise issues of validity, then it would be a way of undermining the system of allocation of jurisdiction under the recast Brussels Regulation in ways which the courts have striven to prevent;
(v) the Arrow case itself had therefore been wrongly decided.
16. The court dismissed all these objections to the grant of Arrow declarations. At [93] the court explained that the existence, following grant, of the statutory remedy for revocation (and, I would add, declarations of non-infringement) needed to be borne in mind. The course envisaged by the Patents Act is that parties should wait and see what patent is granted before resorting to Arrow declaratory relief. However the statute did not create a bar to the grant of relief "in appropriate cases". At [98], giving the judgment of the court which included Longmore and Kitchin L.JJ., I said:
"We have said enough to explain why we do not consider that there is any issue of principle which prevents the granting of Arrow declarations in appropriate cases. Drawing the threads together:
(i) A declaration that a product, process or use was old or obvious at a particular date does not necessarily offend against section 74 of the Act.
(ii) Such a declaration may offend against the Act where it is a disguised attack on the validity of a granted patent.
(iii) Such declarations do not offend against the scheme of the EPC or the Act simply because the declaration is sought against the background of pending divisional applications by the counter-party.
(iv) On the other hand the existence of pending applications cannot itself be a sufficient justification for granting a declaration.
(v) Whether such a declaration is justified depends on whether a sufficient case can be made for the exercise of the court's discretion in accordance with established principles."'
- Biogen also pointed out that the principle that the Court must not usurp the function of the EPO was emphasised in the Arrow case itself (Arrow Generics Limited v Merck & Co. Inc. [2007] EWHC 1900 (Pat)) per Kitchin J at [60]:
""[Merck] says this court should not be making declarations in respect of the validity of patent applications because they are subject to examination by the EPO and their claims can change. For the court to start anticipating the examination process would be to usurp the function of the EPO and this is inconsistent with the framework of the EPC and the Act. I agree with all of these submissions. I find it hard to conceive of any circumstances in which it would be appropriate for this court to grant a declaration that no valid patent could be granted on a divisional application which is being prosecuted before the EPO. But that is not what is sought. Arrow only seeks declarations that its own product was obvious at the priority date."
- At [30] of Glaxo v. Vectura, Floyd LJ also observed:
"There is no dispute that the declaration must be formulated with clarity. The facts ultimately declared by the court must be clear, otherwise the declaration will simply give rise to further dispute and defeat the purpose for which it is granted. The declaration must also be clear so that the court can know what technical issues it has to decide. The declaration must therefore identify the combination of features of the products and processes in question on which the assessment of obviousness is to take place."
- Against this backdrop, Sandoz relied on the widest possible principle from Fujifilm, contending simply that the Court may exercise its discretion to grant declaratory relief when it is useful to do so, citing Fujifilm at [58]-[60]. It may be noted that those paragraphs cite CPR40.20, Messier-Dowty and Financial Services v Rourke, i.e. the most basic principles underpinning declaratory relief in general and some way away from any of the issues raised by Arrow relief, let alone the relief sought in this case.
- It is unnecessary to set out what occurred in correspondence between the parties' solicitors which led up to the claim for the relief I have set out above, because I accept Sandoz's submission that Biogen plainly intend or intended to assert the New Divisional (and any others which may emerge) against Sandoz and its assay if or when it grants. Indeed, it stands to reason that Biogen have been or will attempt to frame a claim or claims in any divisional so as to catch the Sandoz Assay.
- On that basis, Sandoz submit they will be left in a state of continuing commercial uncertainty (even if they are successful in revoking this Patent) due to the presence of the New Divisional (and any others which may emerge). Sandoz therefore submit that that uncertainty can be removed by the grant of the declaration I have already set out.
- Sandoz rely on the matters relied upon in support of their case on insufficiency, which I have dealt with above, but applied to the disclosure of the PCT at its various possible priority dates. Sandoz say there is no suggestion that the disclosure of the PCT adds anything which would make it enabling where that of the Patent is not, or that matters are different at any date in the priority year. Sandoz contends that, as a technical matter, if its principal insufficiency argument succeeds as against the Patent, it must follow that the declaration sought is a correct statement. Sandoz submitted there is nothing in Biogen's evidence which would indicate that it is suggesting otherwise.
- Sandoz therefore submitted that Biogen could only be objecting on the basis that (a) the declaration would serve no useful purpose and (b) the grant of the claimed relief would not be a proper exercise of the Court's discretion.
- In terms of utility, Sandoz contended that the declaration would serve a useful purpose for at least the reasons set out in §23 of the Re-Amended Particulars of Claim, in summary:
i) it will provide Sandoz with commercial certainty regardless of what divisional patents may emerge from the PCT;
ii) it will provide Sandoz with commercial certainty much sooner than waiting for the conclusion of EPO and/or UK proceedings on any such divisional patents;
iii) it will provide certainty not only to Sandoz, but also to the NHS, clinicians and patients that Tyruko and the Sandoz Assay may be supplied and used as intended;
iv) it may be of persuasive value in the courts of other EPC member states and/or promote a wider commercial settlement.
- On the second issue, Sandoz referred to Biogen's pleading that the grant of the claimed relief would not be a proper exercise of the Court's discretion: (paragraph numbers added to identify the three arguments):
"The proposed declaration is concerned not with the novelty or obviousness of a product or process independent of any pending patent application, but with what is and what is not enabled by the disclosure of the PCT. That disclosure is the subject of ongoing prosecution in the EPO. Accordingly (1) the claim for [the declaration sought] is a premature attack on the validity of a patent application prior to grant contrary to s.74 of the Patents Act 1977 and/or (2) a collateral attack on proceedings within the EPO and/or (3) would involve the Court improperly usurping the function of the EPO examiner."
- Sandoz submitted that such arguments were considered by the Court of Appeal in Fujifilm and tackled each one (as labelled above) in turn.
- First, the Court considered what was prohibited by s.74. It held (at [75]-[83]) that s.74 only prohibited a person from seeking a declaration of invalidity (without revocation) of a granted patent, or a declaration that was in substance if not in form a declaration of invalidity of a granted patent. It follows that s.74 is not a bar to the grant of the declaration sought in the present case and Biogen's first point fails.
- Sandoz also submitted that the Court of Appeal then went on to consider whether a declaration that could be regarded as a pre-emptive determination of validity of a putative patent would be a collateral attack on proceedings within the EPO or would involve the Court improperly usurping the function of the EPO examiner:
"84. AbbVie's real complaint, as it seems to us, is that the declarations, by asserting that the dosing regimen is old or obvious, are making it clear that a future patent claim to that regimen would be invalid. Accordingly, if AbbVie were to obtain the grant of claims in that form, the resulting patent would have been pre-emptively adjudged invalid. There is therefore, implicit in the Arrow declaration, an inter partes declaration of invalidity of a putative patent, not yet granted, having those claims.
85. Does the scheme of the EPC and the Act preclude a pre-emptive determination of the validity of a putative patent which has not been granted? If so, then FKB must wait and see whether such a patent is indeed granted and avail themselves of the remedies by way of opposition in the EPO or revocation before the national court. That may never happen. In the meantime there will be continuing commercial uncertainty over whether their product will be held to infringe.
86. In our judgement there is nothing in the scheme of the EPC and the Act to prevent such declarations in cases where there is a real justification for their grant. It is necessary to examine quite carefully the ways in which it is suggested that the grant of such a declaration would conflict with that scheme.
87. So far as the EPO is concerned, the following considerations are relevant. Firstly, the declaration has no direct impact on what the EPO can or cannot do in relation to any given application. The EPO will apply its own internal legal order and procedures irrespective of any decision of the national court. Secondly, the court is not being asked to review or adjudicate on past action within the EPO, as it was in Lenzing or Virgin. It is, however, correct to say that the court is being asked to adjudicate on an issue which may arise in proceedings in relation to one or more applications proceeding in the EPO.
88. We do not think that this latter consideration means that the declaration is a collateral attack on the proceedings in the EPO. On the contrary it is an inevitable feature of the scheme set up under the EPC that national courts will have to decide whether combinations of features are old or obvious, and that they will have to do so while possible divisional applications are still pending in the EPO (or indeed in the national patent office). Whenever a national court decides that a claim with features A, B and C is old or obvious in the course of revocation proceedings against a granted patent, it is deciding an issue which may arise in relation to a pending application. That is exactly what happened in the Arrow case itself, when divisionals were prosecuted in relation to essentially the same idea as had been held obvious (albeit in normal revocation proceedings) by Jacob J and the Court of Appeal. Because of the structure of the EPC, the EPO cannot be insulated from findings by national courts which may run contrary to applications which it is in the process of examining. The EPO is considered by the scheme to be capable of deciding its cases in accordance with its own legal order.
89. For similar reasons, the grant of an Arrow declaration is not inconsistent with the abolition of the right to oppose a patent in pre-grant opposition proceedings. The declaration will not prevent the EPO from granting any patent."
- For these reasons, Sandoz submitted that Biogen's second and third arguments fail. In essence, Sandoz said the declaration sought should be granted because it would serve a useful purpose and there are no special reasons why it should not be granted.
- Biogen's first point was that, in the light of the passages from Glaxo set out above, the declaration sought here is not Arrow relief at all. Instead, Biogen characterise the declaration as a statement that the PCT did not enable the Skilled Team to perform a particular process at the priority date. While the Court obviously has a broad discretion to grant declarations, Biogen submitted it does not extend to usurping the role of the patent office. Biogen characterised the relief sought in this case as unprecedented and submitted that Sandoz appear to be attempting to break new ground.
- Biogen submitted this attempt raises three discrete issues:
i) Are there fundamental obstacles to the grant of relief in these novel terms?
ii) If not, would such relief serve a useful purpose?
iii) If so, is the declaration sought technically justified?
- I propose to address each of these issues in turn, after setting out the respective contentions on each of these points.
- Biogen submitted there were at least two fundamental obstacles to the grant of relief in the form sought by Sandoz that do not arise in Arrow cases.
- Biogen's first point was that Arrow declarations do not necessarily offend against the scheme of the Act or the EPC because they are directed to products or processes that exist independently of any pending patent application. Biogen say that does not apply for this declaration. They suggest it is, in substance, directed to the text of a pending patent application (i.e. the New Divisional) and the issue of what that text does and does not enable. Biogen say that if the Court were to rule on that question, it would plainly involve usurping the role of the examiner contrary to the scheme of the EPC, and ruling prematurely on validity contrary to s.74 of the Act.
- In response, Sandoz submitted that Biogen mischaracterise the declaration sought and that the declaration sought by Sandoz is not directed to the text of the New Divisional any more than an Arrow declaration is directed to the text of any pending divisional. The declaration sought by Sandoz relates to the factual question of what the PCT discloses (or does not disclose). Similarly, an Arrow declaration relates to the factual question of whether a product or process was old or obvious at the priority date: Sandoz cited Mexichem to illustrate this point, where the declaration sought related to whether it was obvious in the light of a Japanese patent application called "Inagaki" to use two chemicals, ze and yf, as refrigerants in a mobile air-conditioning unit (see Mexichem v Honeywell [2020] RPC 11 at H2).
- In neither case will the declaration usurp the role of the examiner, so Sandoz submitted, because these declarations are not a declaration that any particular claim that may emerge from the divisional process will be invalid: nor can they be because the parties seeking this form of declaratory relief do not know what claims the patentee may seek to spin out of the divisional process.
- Sandoz said that Biogen's complaint could only have basis if Sandoz was seeking a declaration that the draft claims of the New Divisional are insufficient, but that is not what Sandoz is doing. Indeed, Sandoz cannot do that because it does not know what the claims of the New Divisional will be, let alone what other claims Biogen may seek to spin out of any further divisional applications it makes.
- Biogen's second fundamental issue was certainty, explained in this way. In Glaxo (and subsequently Mexichem), Floyd LJ gave consideration to the detail with which the product or process must be described for the purposes of an Arrow declaration. In Glaxo he concluded at [32] (emphasis added):
"I do not accept that it is clear at this stage that either the general declaration or the PPD declaration is so unclear that it could not be granted. In each case GSK would have to establish that the relevant features were old or obvious at the level of generality at which they are pitched."
- Biogen suggest there is no problem with this precisely because an Arrow declaration is directed not to any pending patent application, but to a product or process that exists independently. No question of claim construction therefore arises.
- Once again, Biogen say 'not so' for Sandoz's proposed declaration. Unlike the obviousness of a defined product, the question of enablement involves considering the text of a patent and asking whether that text enables the skilled person to perform an invention. It is trite that 'invention' for this purpose means claimed invention. Accordingly, Sandoz's proposed declaration must, of necessity, set up a quasi-claim against which the issue of enablement can be judged. That quasi-claim is to the following process:
"...[performing] an assay to determine the anti-JCV antibody titer of a serum or plasma sample from a patient expressed as an index value (nOD) and, from that index value, to determine the level of risk of that patient of developing PML wherein high risk of developing PML corresponds to an index value of >1.5 or wherein a particular risk of developing PML corresponds to any other index value disclosed in the PCT"
- Biogen pose this rhetorical question: how is this quasi-claim to be construed? And suggested there were only two options: in isolation or in the light of the PCT. If construed in isolation, its meaning is not necessarily relatable to any future claim, and therefore likewise the declaration. If construed in the light of the PCT, this only serves to emphasise that the Court is being asked to usurp the function of the examiner, with an attendant risk of different reasoning on both construction and technical issues that would also serve to make the meaning and effect of the declaration unclear.
- For these fundamental reasons, Biogen submitted that the Court should not entertain Sandoz's claim for declaratory relief.
- Responding to the second 'fundamental' issue, Sandoz made two points in response:
- First, Sandoz said it was based on a misunderstanding of the declaration. Sandoz refuted Biogen's 'quasi-claim' argument on the basis that the declaration sought by Sandoz is a declaration of fact as to what the PCT would or would not have disclosed to the Skilled Team at the relevant dates in precisely the same way as an Arrow declaration is a declaration of fact as to whether a particular product or process is old or obvious or, as in Mexichem, a particular document renders a particular process old or obvious at the priority date.
- This was the point emphasised by Ms Pickard in her reply submissions: the declaration is a factual question as to what the PCT discloses. I sought to test that submission by asking whether that was precisely what the EPO has to decide, to which her response was that the EPO only has to decide whether the particular claims being sought in the EP application under consideration are sufficient or not, and that will require them to go back to the PCT.
- Second, Sandoz contended that the approach being adopted by Biogen of treating the declaration as if it was a patent claim is wrong as a matter of principle. The same point was run by the patentee in Mexichem and was firmly rejected by the Court of Appeal at [19]-[20]:
"Some of Mr Speck's submissions, in his skeleton argument at least, treated the declarations sought here as if they were patent claims. In the conventional approach to interpretation of patent claims, the claimed features are treated as the limit of what is required in order to infringe, in the absence of some express indication to the contrary. Additional features in the defendant's product will not avoid infringement. Thus, argued Mr Speck, the declarations in the present case covered a vast array of different products, such as the combination of ze or yf with every known lubricant, or additional refrigerant. It was quite wrong to pre-judge the obviousness of such combinations. Alternatively, if that was not the effect of the declaration, then it lacked clarity.
I do not think it is correct to construe declarations such as those sought by Mexichem as if they were patent claims, so that every conceivable product which could fall within the declaration is being declared to be obvious. Sensibly understood, what Mexichem is seeking is a declaration that the mere idea of using Inagaki's disclosure of ze and yf as a refrigerant in a MAC is obvious. The declaration, being silent on lubricants and other refrigerants, says nothing about whether combinations of the cited refrigerants with such materials are obvious or not."
- Accordingly, Sandoz submitted that there are no "fundamental obstacles" to the grant of the relief sought by Sandoz.
- Biogen went on to submit that, if they were wrong about that, and on the hypothesis that the declaration is technically justified (which Biogen did not accept: see next heading), the next issue is utility.
- Biogen contended that Sandoz's pleading does not explain how a finding in the terms of the declaration translates into protection from any future claim deriving from the PCT. This is not self-evident. Biogen may obtain a different claim that still reads onto the Sandoz Method. What then? Biogen then indicated they would await to see how Sandoz develops its utility arguments before making further submissions.
- Sandoz's response was that Biogen were making a bad point because it is always the case in claims for Arrow declarations that the person seeking the declaration does not know precisely what claims will emerge in the future. But the fact that the declaration sought may not protect against every conceivable future claim, does not deprive the declaration of utility. Sandoz repeated their point that there appears to be no dispute that, if granted, the declaration sought would have utility for at least the reasons set out at §23 of the Re-Amended Particulars of Claim, as quoted above.
- If the proposed declaration is to be construed in the light of the PCT, Biogen said that it is without technical merit in any event for essentially the reasons given in relation to the Patent.
- In response, Sandoz contended that the parties are in agreement that no new technical issues are raised by this claim for declaratory relief, on the basis of the respective submissions I recorded in [639] and [668] above. Accordingly, Sandoz submitted that if they succeeded on their principal insufficiency argument, namely that the Patent does not enable the Skilled Team to produce an assay in which an index value of 1.5 corresponds to the antibody titer and associated risk of developing PML which the Patent sets as the clinical cut-off for high risk, i.e. the antibody titer which produces an index value of 1.5 in the Gen2 assay, Biogen has no technical defence to the claim for declaratory relief. The only issue that arises is whether the relief should be granted.
- Biogen dealt with this claim very briefly in their written closing. Their point was that the authorities relied upon by Sandoz were in respect of a declaration relating to the nature of the product itself, whereas the declaration sought by Sandoz concerned an alleged insufficiency of the parent application. Biogen submitted that the cited authorities were not applicable.
- A key point from the authorities is one made by Kitchin J. in the original Arrow judgment, where he agreed with the submission that 'For the court to start anticipating the examination process would be to usurp the function of the EPO and this is inconsistent with the framework of the EPC and the Act.' In other words, Arrow relief may (because of the discretionary considerations) be appropriate in circumstances where the function of the EPO (in examining and deciding whether to grant EPs) is not usurped. As I understand the authorities, the Arrow jurisdiction bypasses the function and jurisdiction of the EPO and hence no usurpation occurs. Thus, a finding that the alleged infringer's product or process is old or obvious does not involve any decision to be made by the EPO.
- In this case, however, and although the argument was not put in this way by Sandoz, it seems to me on reflection that the effect of the declaration sought would be to put Biogen on the horns of a dilemma:
i) On the one hand, what the PCT disclosed was a division between high and low risk of developing PML based on various possible cut-points in the Gen2 assay.
ii) On the other hand, the PCT did not disclose sufficient information for the Skilled Team to reproduce the Gen2 assay sufficiently to be able to implement the same cut-points.
- In effect, the declaration declares that the PCT cannot give rise to a valid patent, because any patent derived from the PCT would necessarily be insufficient. Even if that is the correct conclusion, it is not a finding that the UK Court can make, unless or until a granted EP is litigated before it. It is for the EPO to decide whether any EP should be granted. In my view, to make this declaration would be to usurp the function of the EPO, whether on the New Divisional or any other divisional from the PCT.
- Accordingly, I consider I must decline to make the declaration sought.
- Furthermore, I bear in mind that the findings I have made in this Judgment on validity (assuming they will remain undisturbed) provide a degree of certainty to Sandoz and, possibly, some ammunition to use in the EPO, although I am certain the EPO are quite capable of determining whether any divisional, if granted, would be insufficient or invalid for some other reason.
- For the reasons explained above, if the Patent is valid, then the method specified in the claims is not used in the UK and so there is no infringement. If the Patent were valid and the method was used in the UK then it would be infringed on an application of the doctrine of equivalents. However, in my judgment, the Patent is plainly invalid. Sandoz are not entitled to the declaratory relief they sought.
- Finally, I wish to point out that the outcome of this case in no way reflects on the excellent science conducted by the group at Biogen which has advanced the understanding of the risks of a horrible condition, PML. If anything, it reflects on the way Biogen and its advisers approached the patent bargain.
